Advances in metabolomics drive a parallel surge in metabolite discovery – and inspire new platforms able to measure increasing numbers of compounds in blood, plasma or urine. Our ability to perform such analyses at the microsample scale – a single drop of blood dried on filter paper – has given rise to modern newborn screening.
Here, I describe how this revolution in metabolomics and clinical diagnostics all started with the analysis of amino acids and acylcarnitines 20 years ago.
Determination of L-carnitine and its derivatives is the basis for newborn screening for several potentially deadly metabolic disorders – a prime example of how analytical science affects all our lives, from the day we are born. L-carnitine is unique and fascinating molecule. With some chemical similarity to amino acids (perhaps even an ancient precursor to amino acids – see “The Primordial Group” below), L-carnitine is a quaternary ammonium compound – a rare class in the human body. It plays a crucial role in fat metabolism, helping to break down fatty acids (through beta oxidation) into a form that the mitochondria – the cell’s powerhouse – can use as fuel. Beta oxidation is facilitated by coenzyme A, but coenzyme A and its acyl-bound fatty acids (acyl-CoAs) cannot always cross the mitochondrial membrane. Short- or medium-chain acyl-CoAs can traverse the membrane, but very-long-chain acyl-CoAs cannot, so they must be temporarily transferred to another substrate for transport across the membrane via a translocase “tunnel.” L-carnitine is that substrate. Figure 1 shows how L-carnitine transports fatty acids through the inner mitochondrial membrane in the form of acylcarnitine. Once inside the mitochondria, the fatty acids are transferred back to coenzyme A for delivery to beta oxidation enzymes. Using a train analogy, coenzyme A can be thought of as a small engine moving a few train cars at a time around a rail yard, while L-carnitine is a bulkier locomotive that can pull longer trains over greater distances.
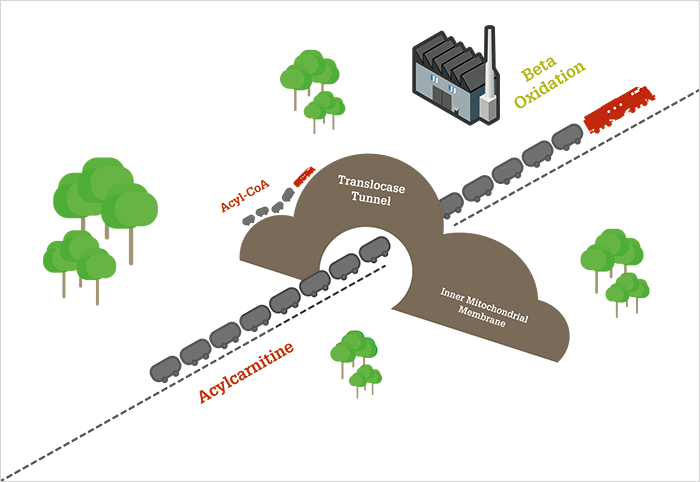
There is an additional role for L-carnitine: if an excess of acyl-CoAs has accumulated in the mitochondria, it will transport these out into the cell and onward to the blood for elimination. And that’s why a metabolic disorder of beta oxidation can lead to a deficiency in L-carnitine – if certain fatty acids cannot be metabolized they build up in the mitochondria and have to be eliminated as acylcarnitines in urine or bile, leading to gradual loss of the “engines” that deliver the fatty acid cargo. Such disorders are highly unusual but very dangerous. Provided there is sufficient glucose to fuel metabolism, fatty acids simply accumulate, causing fatty liver but often no outward signs of toxicity; but if blood sugar drops, there is no “safety net” in the form of fat metabolism. Muscle weakness followed by coma and death can progress rapidly and without warning. These tragic consequences can be avoided relatively easily with diet and medication, but by the time symptoms occur, it may be too late. Both the understanding of the biochemical role of L-carnitine and also its potential in clinical analysis evolved rapidly and in parallel.
Which came first, the chicken (disease) or the egg (method)?
L-carnitine is a powerful example of what I like to call the clinical chemist’s feedback loop: technology changes medicine, which changes technology. Analytical chemists in the 1980s were faced with a problem – L-carnitine and acylcarnitines had great potential as markers of fatty acid metabolism, but these non-volatile compounds could not be analyzed using gas chromatography-mass spectrometry (GC-MS) – the most common tool for metabolic research of the time. To overcome this hurdle, they developed complex sample preparation procedures to remove the quaternary amine and make L-carnitine volatile for GC, while keeping the molecule structurally informative. But these methods were limited to measuring free and total L-carnitine; it was impossible to identify individual fatty acylcarnitines. Consequently, there was still relatively little data on acylcarnitines until the mid-1980s, when significant advances in LC-MS interfaces were made (thermospray and liquid secondary ionization methods, such as fast atom bombardment, FAB). As L-carnitine is a preformed cation with at least one positive charge in aqueous solution, it is the perfect substrate for positive-ion LC-MS.With further MS advances and the availability of less expensive tandem MS (MS/MS) systems, clinical investigations expanded, and the role of carnitine in fatty acid metabolism was better understood. Clinical investigators studying infants with inherited disorders in fatty acid metabolism had a new and better analytical tool, and were quick to make use of it. Using analytical data from urine, plasma and blood analysis and numerous biomarkers from organic acids to acylcarnitines, it was possible not only to understand mitochondrial fatty acid oxidation but also to develop a test to detect beta oxidation disorders shortly after birth, while they are still treatable. Ultimately, the technology set the stage for an important and significant expansion of newborn screening. Early MS/MS used cumbersome ionization techniques (FAB or fast ion bombardment, which require a difficult insertion and manual analysis) but, soon after, new ionization methods were established; for example, electrospray, which enabled automation and easier population analysis. Today, pathology laboratories in most developed nations screen infants using MS/MS. Even now, the technology is still being expanded to include new diseases – spawning even more advancements. In my view, it is this feedback loop (Figure 2) that makes analytical science key to discovery and better health.
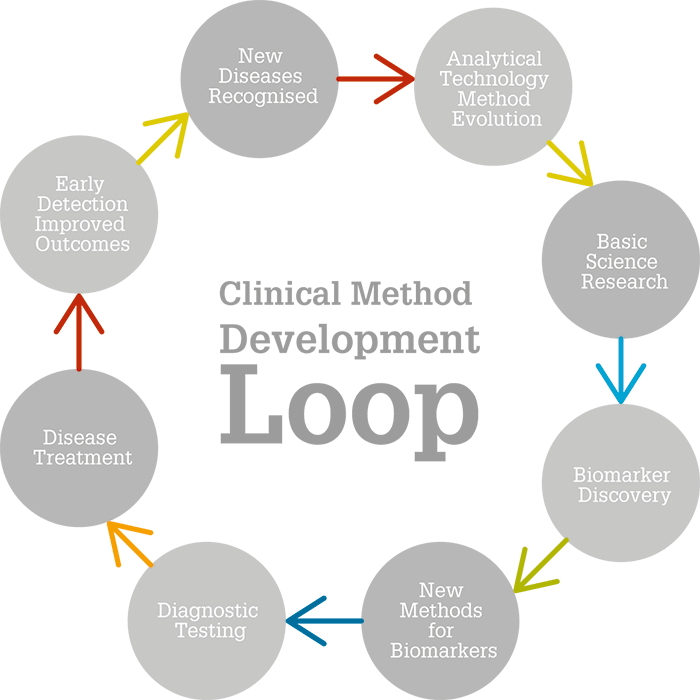
A revolution in newborn screening
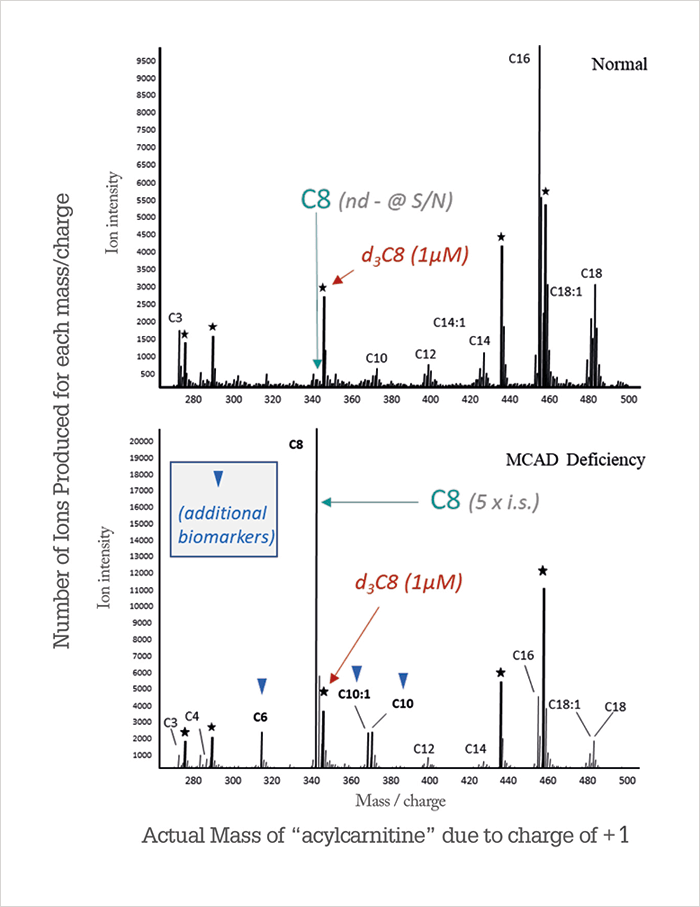
The fundamental problem with most clinical diagnostic tests is that you only test once symptoms appear, so you run the risk that the disease will have already caused irreversible damage or even death before treatment can be started. Still, in the technology loop described earlier (Figure 2), it is a necessary step prior to developing a screening technique and is extremely important in confirming disorders detected by screening. Information from the metabolic profiles of a diagnostic test tells us what to look for in developing early screening. As we uncovered beta oxidation disorders in sick patients, in unexplained deaths, and from family history, it became obvious that a newborn screening test could save many lives, especially as treatment is often simple and noninvasive – a low-fat, high carbohydrate diet (a literal “spoonful of sugar”) and supplemental L-carnitine.Though the treatment for fatty acid disorders may be simple, the evolution of newborn screening of acylcarnitines was anything but. Tandem MS was a game changer when it was introduced in the 1990s, but it took at least another 10 years for its use to be accepted and widely implemented for newborn screening. One of the first MS/MS screening tests was developed for MCAD deficiency. A genetic survey for the most common gene associated with the disorder showed that the frequency of the mutation was 1 in 10,000–15,000 (a similar frequency to phenylketonuria, a disorder of phenylalanine metabolism that was the first disease screened in newborns more than 50 years ago). MCAD deficiency is a disorder of the first steps (dehydrogenation) of beta oxidation for medium-chain (6–12 carbon units) fatty acids; if the enzyme is deficient (or defective), the oxidation of these fatty acids does not occur and precursors accumulate in the mitochondria. The precursors are coenzyme A adducts octanoyl (C8), hexanoyl (C6), decanoyl (C10), and decanoyl (C10:1), which cannot be measured directly in the blood because they are never transported out of the mitochondria. As mentioned above, one of the roles of carnitine is to remove excess acyl-CoAs from the mitochondria (to keep it functioning) and transfer them to blood for elimination in urine and bile; thus, we can measure acylcarnitines as a substitute for acyl-CoAs. In MCAD deficiency, we observe elevated medium-chain acylcarnitines in a unique pattern, with all four peaks clearly increased as shown in Figure 3. In normal metabolic states, C8 concentrations are barely distinguishable from noise; in MCAD deficiency, it often becomes the most significant peak in the mass spectrum.
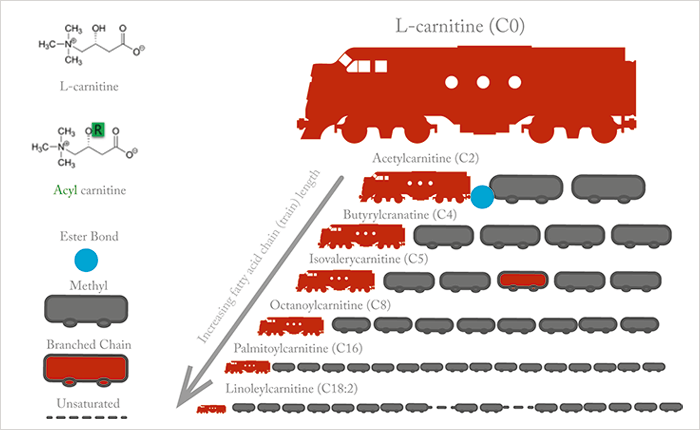
What is the role of L-carnitine in fatty acid metabolism? And what happens when things go wrong? The biochemical role of L-carnitine is to transport very-long-chain fatty acids across the inner mitochondrial membranes into the mitochondrial matrix. It might be helpful to imagine that L-carnitine is the engine (locomotive) that transports its cargo (oil tankers/fatty acids) into the mitochondria. Figure 4 illustrates this “train” analogy from a structural perspective.
The mitochondria are the power houses of metabolism and ATP generation. They are also the site of beta-oxidation of fatty acids. Figure 5 illustrates the metabolic pathways of fatty acids and the role of L-carnitine and acylcarnitines. Note the large number of enzymes and proteins involved in transporting fatty acids across the inner mitochondrial membranes and processing them once inside – at each of these enzyme or protein steps, a defect in the protein will cause metabolic disease. As in most metabolic disorders, the metabolite normally processed by the defective enzyme or protein accumulates, while metabolites further down the pathway may decline.
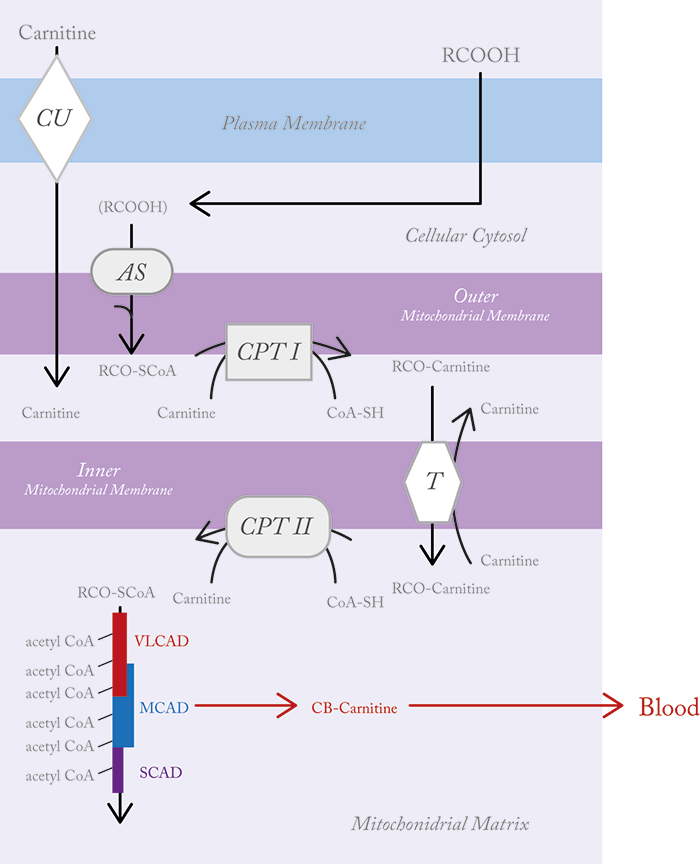
Take a disorder in CPT II (carnitine palmitoyl transferase type II), the enzyme responsible for transferring the fatty acyl group from the long-chain acylcarnitine back to acyl-CoA – the substrate involved in beta oxidation. When CPT II is defective, acylcarnitine is not released to acyl-CoA and accumulates in the membrane and inner mitochondria; short chain acyl-CoAs are not produced and hence beta oxidation declines. Only those acylcarnitines that are small enough to penetrate the membranes without the assistance of carnitine are metabolized (see Figure 1). Blood tests in those with the disorder show excess long-chain acylcarnitines with very low L-carnitine because it is all bound up in the long-chain acylcarnitines. This high ratio of long-chain acylcarnitines to L-carnitine is the basis for detection of a metabolic disease by MS/MS.
Before newborn screening, a diagnosis of MCAD deficiency required either urine organic acid analysis or a repeat acylcarnitine profile after intravenous L-carnitine supplementation. Why? Acylcarnitine formation clearly depends on the availability of L-carnitine, but as beta oxidation defects (such as MCAD deficiency) deplete L-carnitine, the concentrations of these metabolites decline. Fortunately, L-carnitine is not generally deficient at birth so metabolites are easily detected in newborns. The use of acylcarnitine profiles in newborn screening has saved the lives of many children born with inherited disorders. Such screening shows the power of metabolomics in practice – there is no better example of how clinical research can evolve to become standard practice. But what about alterations of metabolism not due to inherited disease?
Beyond newborn screening
In my opinion, detection of inherited disorders is just one piece of the jigsaw. In particular, measurement of acylcarnitines, together with amino acids, may be important in understanding metabolism, growth and nutrition in premature infants. Unlike detection of inherited metabolic disease, changes in metabolism in preterm infants are temporary and result from indirect factors, such as immature organs. The neonatologist’s toolbox for metabolic investigations has historically been limited by a reluctance to move away from older “tried and tested” technology, the need for rapid turnaround (an HPLC amino acid analysis or urine organic acid could take several days) and, most importantly, the amount of blood that can be safely taken from an infant who may weigh less than 1 kg. However, better metabolic tests are sorely needed in these smallest of patients. For example, preterm infants require extra protein compared with a full-term baby, to mimic the high level of amino acids they would be exposed to in the womb. The gut of a 22–28-week-old premature infant is not able to extract the protein they need from their diet, so they require intravenous amino acids (parenteral nutrition) to provide the building blocks for growth. The challenge is knowing how much of this amino acid mixture to administer to provide the fuel for growth – too often this is based on guesswork.By now, you may be asking yourself – what does this have to do with L-carnitine?
In full-term newborns, we have observed that acylcarnitines decline rapidly after birth, stabilizing at 1-week-old to near-adult levels. It is thought that after the stress of birth and the sudden “cut off” from nutrition provided by the mother via the umbilical cord, newborns rely on fatty acid metabolism until they switch to a steady glucose supply in the form of milk. Regardless of the cause, acylcarnitines decline, but L-carnitine is relatively stable. In preterm infants, acylcarnitines also decline but free L-carnitine levels depend on the actions of their neonatologist. Modern bioinformatics have revealed two distinct populations of preterm infants: those who received L-carnitine as part of their IV nutrition and those who did not (1). The infants receiving supplemental L-carnitine had high levels of free L-carnitine, as well as higher acylcarnitine concentrations. Infants who were not administered L-carnitine saw levels decline over the first week of life until they were close to a deficiency. In all cases, once infants moved from parenteral nutrition to feeding normally, L-carnitine increased back to near-normal levels. Because the level of acylcarnitines depends in part on the availability of L-carnitine, it can create confusing results from an analytical point of view. For example, monitoring the catabolic product of leucine, isovaleric acid (which forms an acylcarnitine), is unhelpful because its levels may be higher or lower depending on the levels of L-carnitine. Further, organic acids for amino acid catabolism and fatty acid catabolism merge in beta oxidation. Lipids are also supplemented in parenteral nutrition, and high doses of both amino acids and fatty acids can create a perfect storm of metabolites in beta oxidation and overflow the system, causing toxicity. Currently, neonatologists monitor ammonia but this only reflects amino acid catabolism and not fatty acid oxidation. The solution is to normalize all levels of acylcarnitines to L-carnitine and that is currently being investigated from a bioinformatics perspective.What’s next for L-carnitine?
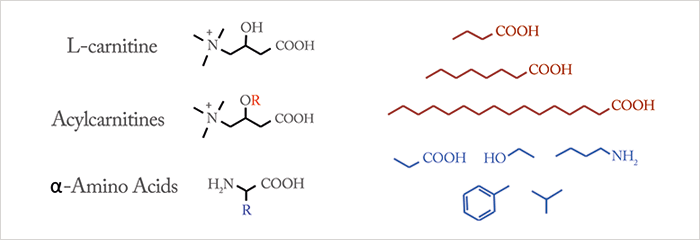
L-carnitine is still not well understood beyond its role in fatty acid metabolism and the obvious problems associated with a deficiency. That doesn’t stop it being widely sold as a supplement in the vitamin aisle – previously called Vitamin BT (it is not a vitamin), it is used frequently in body building and sports for the perceived benefit of metabolizing fat more efficiently and improving muscle function. These claims are dubious, at best. But could L-carnitine have qualities we are not yet aware of? With the detection of hundreds of infants with rare metabolic disorders, the prophylactic administration of L-carnitine became a topic for debate. The rationale behind using L-carnitine for metabolic disease is to correct a deficiency and by doing so keep beta oxidation functioning more efficiently, especially at times when fats are burned faster or glucose is used more quickly. Diseases like MCAD lead to acute illness when glucose becomes depleted – the reasoning is that supplemental L-carnitine can keep the dysfunctional fatty acid oxidation functioning slightly longer and better, until glucose can be replenished. It is also believed to help keep muscle functioning more efficiently in terms of fat metabolism, preventing or reducing cardiomyopathy in some metabolic states. It is not difficult to see how the scientific and medical use of L-carnitine has been translated into the pseudoscientific claims made by supplement companies. It seems unlikely that L-carnitine will have a significant impact on muscle function in healthy adults, who produce more than enough L-carnitine for their needs. However, it’s not inconceivable that there may be roles for L-carnitine in metabolic states that are highly reliant on fat metabolism, including brain biochemistry and disease such as stroke. I wouldn’t be surprised to see this multifaceted molecule reimagined once again, as new roles and functions are discovered. Donald H. Chace is Chief Scientific Officer, Medolac Laboratories, Lake Oswego, Oregon, USA.Could L-carnitine provide clues into our biochemical evolution? A scientist at a major pharmaceutical company once told me that L-carnitine should be thought of as a “prehistoric” amino acid. The idea of L-carnitine as a primitive amino acid immediately appealed to me for several reasons. First, both amino acids and L-carnitine contain basic amine and carboxylic acid functional groups, which become charged (zwitter ions) in water. Both amino acids and L-carnitine have a central carbon that separates the amino and carboxylic acid functional groups. This central carbon also contains a variable “R” group that changes the molecule both structurally and functionally. Figure 6 shows “generic” structures for L-carnitine, acylcarnitines and α-amino acids. We assume that early life (in the primordial soup) required more reactive functional groups as pH and temperature conditions were widely variable in Earth’s early history. With time, environmental conditions and pH stabilized and more efficient metabolic processes evolved, catalyzed by enzymes. The idea of carnitine as a “prehistoric” precursor to amino acids comes from the strong ionic nature of the quaternary amine (four carbons attached to nitrogen), making it permanently charged in aqueous solution regardless of pH. By comparison, amino acids are primary amines (one carbon attached to nitrogen), and are only charged in aqueous solution at physiological pH (7.4). To further this “ancient history” concept for L-carnitine, it is worth noting that it has a unique role in our most ancient organelle, the mitochondrion – among other more important functions like energy production, the mitochondria house our ancient and conserved DNA. The evolution of mitochondria and role of ancestral DNA has attracted attention recently, and thus it is quite natural to wonder about the evolutionary role of L-carnitine and why it plays a vital role in mitochondrial fatty acid metabolism.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- RH Clark et al., “Impact of L-carnitine supplementation on metabolic profiles in premature infants”, J Perinatol, 37, 566-571 (2017).



