Aerosols play an undeniably important role in atmospheric processes. And our understanding of that role has become increasingly clear over several decades. Initial research focused on inorganic aerosols, in large part because they posed questions that we thought could be answered with established methods. Sure enough, we began to answer some important questions – but, as is typical in research, some answers inspired new questions that pushed the limits of what was then current analytical science. It soon became clear that bulk chemical measurements with time scales of days to weeks would not yield the information necessary to advance the state of knowledge with respect to aerosols. We had several important yet unanswered questions; for example, how important is the chemistry of single particles? What is the role, if any, of organics? Aerosol mass spectrometry was developed with these new questions in mind and has been revolutionary in providing tantalizing glimpses into the complex life cycles of aerosols. However, it has fallen short when it comes to organics. Questions still remain: How do we untangle the molecular level chemistry of organic aerosols? Do we even need to? What methods exist that could give us information to this end? Unfortunately, the answer to the last question is “none.”
I would say that, for the scientific community in this field, complacency is a major enemy. Because we are able to produce some data with aerosol MS – data that can easily be handled by models and used to gain some predictive model outputs – many of us simply stopped looking for better data. And indeed, the general thinking has become: “why bother getting more detailed data when the models can’t handle it anyway? Let’s just work with what we have.” I wasn’t – and am still not – satisfied with that conclusion...
More lasers, more fun
My fascination with aerosols actually started at the same time as my career in academia, some 20 years ago. I was trained as an analytical chemist at the University of Toronto before entering a PhD program at the University of Florida. I was under the mentorship of the brilliant James Winefordner where I learned to appreciate the power of lasers (pun intended). In fact, my motto has since been “more lasers, more fun.” Shortly before completing my postdoc, I received an NSF Fellowship that allowed me to continue my work at the Joint Research Center (JRC) of the European Commission in Ispra, Italy. And in late 1993, I had the good fortune to work with another great mentor – Nicolo Omenetto, who I first met in Florida during one of his annual summer pilgrimages to the Winefordner group. The fellowship proposed a continuation of my PhD work on the use of two-step resonance-enhanced laser ionization as an ultrasensitive, monochromatic photon detector capable of operating effectively in bright light conditions. And what a project that was – two lasers, a low-pressure lamp and a voltage-to-frequency converter that whistled when the laser wavelength was tuned!It was toward the end of my fellowship that I met an energetic and overly-enthusiastic German postdoc (Ulrich Panne, now President of the Federal Institute for Materials Research and Testing) who had a contagious excitement for all things aerosol. In fact, he had joined our lab to initiate an aerosol laboratory – in keeping with one of the focuses of the Fourth Framework Programme of the European Commission. We were tasked with developing a new instrument for the chemical analysis of atmospheric particles. The analysis needed to be accomplished on a single particle basis, in real time and in situ – a daunting task. There were several groups worldwide working toward the same goal and, without exception, all approaches used mass spectral detection of ions generated by one of several different means. Most of the approaches used high-power lasers to intercept the particles in the ionization region of the mass spectrometer. During this laser-particle interaction, copious numbers of ions were generated for analysis. At the time, all the groups were focused on the analysis of inorganic particles. Of the groups working on this task, two produced commercial instruments – one of which has been revolutionary in this field (the Aerodyne aerosol mass spectrometer). The innovative, plug-and-play approach presented a paradigm shift in the analysis of atmospheric aerosols. It was accessible to a large number of research groups and provided, for the first time, a glimpse into the chemical makeup of organic aerosols (OA) primarily in the form of oxygen-to-carbon ratios (O:C). The approach quickly gained a strong following among the atmospheric chemistry and physics communities, leading to an expansive user network across the globe. Its impact in the field cannot be overstated. And yet, despite the advances made possible by this instrument, something inside me still continued to ask: “Can something as simple as O:C really paint a meaningful picture of the role of OA in global change?” As chemists, should it not be “obvious” that distinct molecules are important? And that chemistry fundamentally governs the behavior of OA in the atmosphere? It was my belief that to advance our understanding of the governing principles of OA chemistry and atmospheric impact, we must push aside the naivety or complacency of our field.
But how could we approach the task of analyzing femtogram quantities of molecules on/in OA without destroying the molecular identities of the compounds? And how could we accomplish this goal while working at atmospherically relevant mass loadings and time scales? At the JRC, we also developed an aerosol mass spectrometer based on a relatively high-power pulsed laser that was capable of measuring positive and negative ions simultaneously. Although we anticipated observing predominantly positive ions, we were surprised to measure negative ions with greater resolution and even better sensitivity. Unfortunately, our negative ion spectra were highly fragmented and much of the molecular information was lost. Nonetheless, we were excited at the prospect of making nascent OA measurements.
The birth of NIR-LDI-AMS
In mid-2000, my stay at the JRC ended and I took up my current faculty position at the University of Vermont. I started to think about ways we could be gentler in our vaporization and ionization approaches, so that we could reduce – or ideally eliminate – molecular fragmentation. Was there a way of keeping molecules intact, while still maintaining the sensitivity needed for measurements at ambient levels? My original approach was to use infrared laser vaporization with resonance-enhanced multiphoton ionization, harking back to my PhD studies (“more lasers, more fun”!). Unfortunately, the funding-powers-that-be made it clear that they thought my approach was quite limited, while others expressed skepticism of the need to measure organic aerosols at all. “After all,” they wrote, “how much organic mass could there really be in PM2.5?”At this point in my life, I started to question my career choice! Fortunately, while describing the photoelectric effect to my general chemistry class at UVM, I remembered an observation from my JRC work about the measurement of negative ions. Could it be that the particles were acting as little sources of photoelectrons, which then attach to vaporized molecules? I rushed back to my lab right after class and discussed this idea with my graduate student (Brian LaFranchi) and within a day or so, we had modified our aerosol mass spectrometer to use a home-built thermal vaporizer and a photoelectron source for testing. It took more than three weeks of constant fiddling to record our first spectrum of pure particles of oleic acid that showed absolutely no measureable fragmentation; and we were off to the races... But after several years of studying the ionization method of photoelectron capture, we accepted that it would never provide us with the necessary analytical figures of merit to work at ambient levels. Now what? My first thought was to couple the photoelectron source to laser vaporization of the particles. In this way, we could optimize the temporal overlap between the vaporization laser and the UV laser generating the photoelectrons. A simple, back-of-the-envelope calculation suggested that, if we vaporized from a small collection probe in the vicinity of the photoelectron source, we could get signal enhancements on the order of 1,000 – 10,000 fold. Once we had incorporated laser vaporization from a small 1-mm diameter pure magnesium probe, we were rewarded with an approximately 3000-fold increase in ion signal, still with no molecular fragmentation! For once, a “rationally thought-out mechanism for improvement” worked as expected – that is, until we realized that the UV laser was not firing, which meant that the near-IR pulse was solely responsible for the signal enhancement! Of course, I was elated that this meant that the complexity of the instrumentation would be reduced greatly. But I will confess that I was disappointed to have been wrong once again. In fact, the addition of the UV pulse in this instrumental configuration provided no substantial benefit to using the near-IR pulse alone. And thus, near infrared laser desorption/ionization (NIR-LDI) aerosol mass spectrometry (AMS) was born.
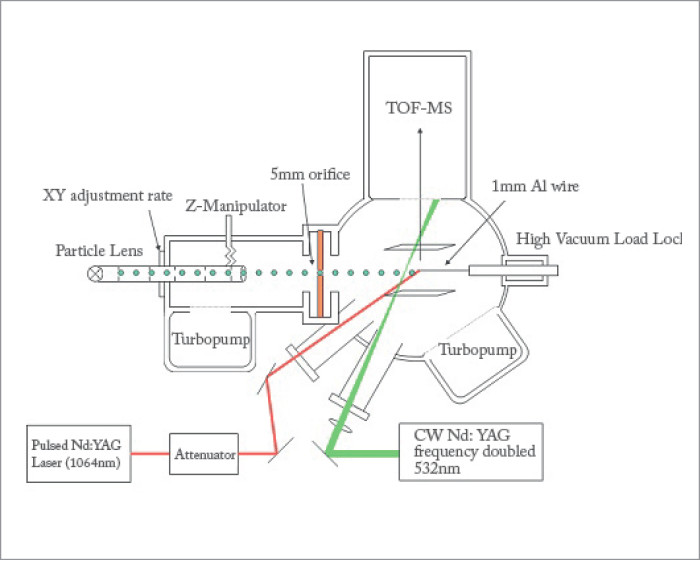
Simplifying the Puzzle
One could liken the difference between hard- and soft-ionization mass spectrometry to the difference between the frustration of assembling a 1000-piece puzzle after several glasses of wine (and without a copy of the final picture), and the simple enjoyment of helping a toddler put together a 50-piece floor puzzle while enjoying your first glass of wine... In our instrument, aerosols are sampled via an aerodynamic lens to produce a tightly collimated particle beam that is directed onto a metal probe in the ionization region of the mass spectrometer. After some suitable collection time of seconds to minutes (depending on the organic aerosol mass loading), the NIR laser is fired, generating a 4 ns burst of ions that are chemically analyzed by time-of-flight mass spectrometry. The power density of the laser at the probe surface (<10 MW cm-2) is below the plasma formation limit and most organic molecules are transparent to the wavelength we use (1064 nm). Furthermore, even for those few photons that may be absorbed, the photon energy (~1 eV) is not sufficient to photoionize organic molecules directly. The net result is that all molecules are measured as their intact, pseudo-molecular ions ([M-H]-), greatly simplifying interpretation of the chemical state of the particles.The physical state of particles can affect particulate phase chemical reactions, and thus the growth rates of newly formed atmospheric particles. The phase state may also markedly increase the lifetime of organic aerosols, as the oxidation caused by atmospheric ozone and other oxidants is confined to the surface. Recent results suggest that biogenic SOA particles can adopt a solid, and most likely glassy state. Furthermore, it has been shown that for mixed compositions of isoprene and alpha-pinene derived SOA, a significant number of particles bounce off (rather than adhere to) solid surfaces, suggesting a fraction of the particles are not in the liquid state.
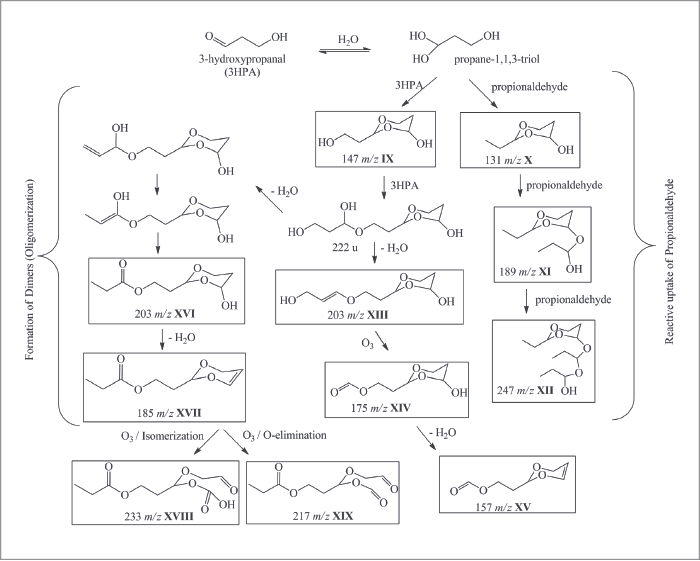
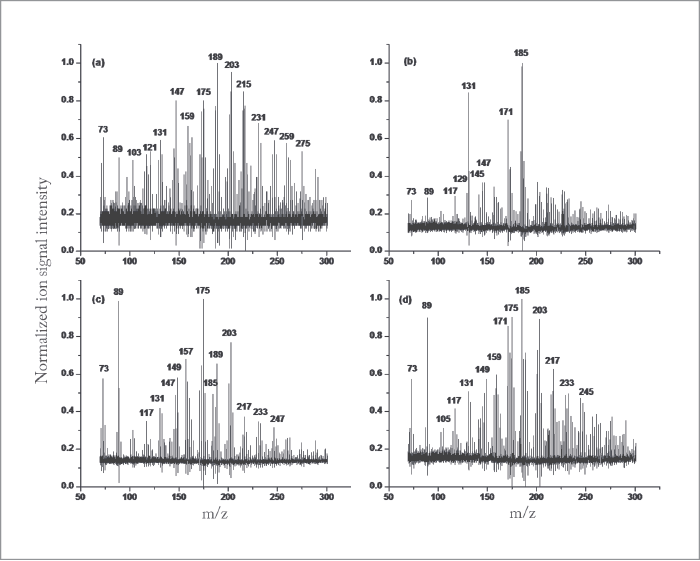
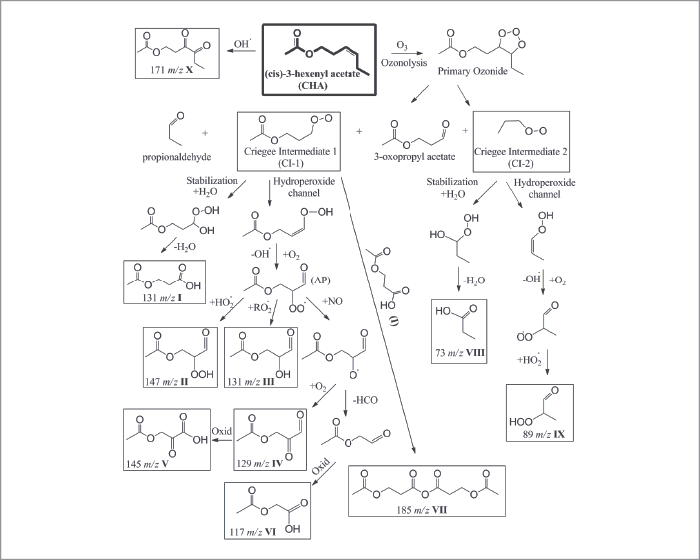
Of course, our original experiments were conducted with pure organic particles, such as oleic acid, to characterize the analytical figures of merit of our approach. We then reacted these oleic acid particles with ozone in a flow reactor, to demonstrate the exceptional capability of our method in deconvoluting the heterogeneous oxidation that takes place as ozone molecules are reactively used by the oleic acid particles. It should have been a simple undertaking, with only four chemical products predicted from homogeneous chemistry in the bulk (actually, a beaker). Basically, ozone will add electrophilically across the double bond of oleic acid, cleaving it to produce four oxidization products of lower molecular weight. Imagine our surprise when we measured not only tens of oxidation products of lower molecular weight, but also products of much greater molecular weight than the parent oleic acid molecules. What was going on? We spent the next several years trying to understand this heterogeneous chemistry, and it turns out that the reaction of even a simple, one component organic particle was full of surprises: stabilized Criegee intermediates participating in multigenerational chemistry, leading to oligomers, formation of extensive hydroperoxide networks and high concentrations of volatile products measured in the particle phase, to name but a few. Clearly, our picture of heterogeneous aerosol chemistry was far from complete. Our next step was to measure secondary organic aerosol (SOAs) formed by oxidation of common and extensively studied terpenoid compounds emitted primarily by tree foliage. The experiment that has become a “rite of passage” for anyone working in this field is the ozonolysis of α-pinene. During these first experiments, we noted that, after 20-30 minutes of aging in our environmental chamber, the SOAs no longer produced any measurable ion signals in our NIR-LDI-AMS. We conducted a similar experiment with limonene as the SOA precursor and saw the same behavior, albeit after more than 45-60 minutes. After much effort trying to determine the loss of signals, we thought perhaps that the SOA particles were not liquid (as has always been assumed), but rather were turning “solid” and bouncing off the probe surface upon impact, rather than being captured. Of course, it is a difficult to make the case that “the particles must be changing phase because we lose our signal,” so an independent measure of the particle phase was required. Once again, no method existed for the on-line, near real-time measurement of the particle phase in a polydisperse aerosol and we found ourselves in the “exciting situation” of having to develop a method to make measurements that were previously not possible. Building on the work of Annele Virtanen (now at the University of Eastern Finland) and her group on the use of particle bounce to infer particle phase (1), we modified an electrostatic low pressure impactor (ELPI+) to use, in turn, impaction plates that either promoted or eliminated bounce (2). Using this approach, it was readily apparent that the phase-state of SOA changes rather dramatically over short time periods, supporting our earlier observations of the loss of ion signals from the NIR-LDI-AMS.
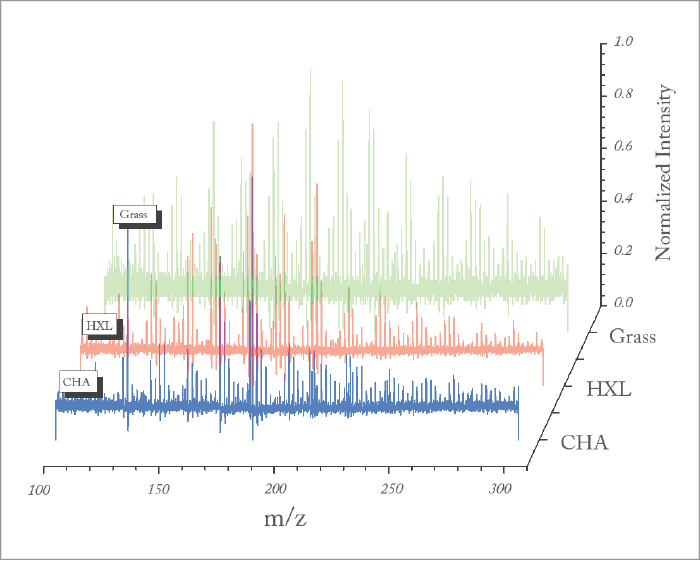
Understanding SOAs
Ultimately, the question arises whether the ability to measure particle phase and particle chemistry at the molecular level is truly needed to better understand the formation, aging and role of SOA in our atmosphere. For example, is our original hypothesis that distinct molecular products are important in defining the optical properties of SOA supported by these new data? Having taken advantage of the analytical capabilities of NIR-LDI-AMS to measure the chemical composition of SOA at the molecular level, we were able to identify new SOA precursors emitted from turfgrass. These compounds, termed green leaf volatiles, or GLVs, were dominated by two reactive compounds: cis-3-hexenol (HXL) and cis-3-hexenyl acetate (CHA). Interestingly, we were able to distinguish that ozonolysis of each compound proceeded via different chemical pathways. HXL proceeded via an oligomerization pathway that leads to greater concentrations of higher molecular weight products than was the case with CHA, which proceeded through a hydroperoxide channel. The difference in reactive pathways bore itself out in the measured bounce behavior, with the HXL-SOA exhibiting much greater bounce (which is to say, higher viscosity). It is interesting to note that the bounce behavior of the SOAs were amplified more than ammonium sulphate, which is used as a reference solid aerosol. What is one to make of that? Such time dependent phase behavior makes treatment in models difficult at best. On the question of optical properties, it has become common to relate particle optical absorption and scatter with the average O:C ratio of the aerosol, with higher O:C indicative of greater optical absorbance. However, this approach can be misleading in some cases. For example, in collaboration with Scot Martin and his group at Harvard University, we generated GLV SOA in a continuous flow environmental chamber, all the while monitoring O:C ratio with an Aerodyne AMS, chemical composition with our NIR-LDI-AMS and optical scatter with a nephelometer. For the two SOA precursors under low relative humidity (~10 percent), while the CHA SOA has a lower O:C, it exhibits a stronger absorbance than HXL SOA. Similarly, when comparing the absorbance of HXL SOA at 10 percent and 70 percent relative humidity, the SOA formed under humid conditions has a significantly greater absorbance, despite indistinguishable O:C for the two systems. In the former case, comparison of the NIR-LDI-AMS particle mass spectra for SOA formed from each precursor clearly show very different chemical fingerprints. In fact, if we calculate the carbonyl content (that is, the area of all ion signals known to originate from carbonyl-containing compounds normalized to the summed area of all ion peaks), we see that CHA SOA has a carbonyl content of 3.5 (±0.8) percent, as compared to 2.1 (±0.5) percent for the HXL SOA, indicating a higher predicted absorbance for CHA SOA.The punchline
Our molecular level analyses have given us insights into the life cycles of organic aerosols that were entirely new and, in some cases, completely unexpected. We’ve developed instruments and made measurements that have allowed us to greatly advance our understanding of the life cycles of organics in the atmosphere. Our new question: “can this knowledge be used to help understand the roles and quantify the impact of organic aerosols in our atmosphere?” Box models have made great strides in their ability to make these predictions. However, owing to still-limited computing power, we must make substantial assumptions and simplifications. With the availability of molecular-level data, the challenge we now face lies in incorporating these fundamental data to generate models that, although more complete and accurate, are computationally unfeasible. Given the current computational constraints, what are we willing to sacrifice within these models to include this new chemical understanding? Time will tell. Giuseppe Petrucci is Professor in the Department of Chemistry at the University of Vermont, Burlington, USA.Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- A Virtanen et al, “An amorphous solid state of biogenic secondary organic aerosol particles”, Nature, 467, 824-827 (2010) S Jain and GA Petrucci, “A New Method to Measure Aerosol Particle Bounce Using a Cascade Electrical Low Pressure Impactor”, Aerosol Sci. Technol. 49, 390-399 (2015).



