A new era of medicine
Recent advances in gene therapy – such as CRISPR/Cas9-based tools – are allowing researchers to introduce or modify genes with unprecedented feasibility. These technologies are hugely exciting from a therapeutic standpoint, particularly in addressing rare and chronic diseases. The ability to potentially heal lethal diseases based on genetic variants, such as muscular dystrophy, cystic fibrosis, and Huntington’s disease, explains the hype around these technologies. In addition, the versatility of gene therapy approaches seems endless.
Diabetes, a chronic disease affecting over 400 million people globally according to the WHO, is another prime example of how potential therapies could be life-changing – I would know, I am diabetic myself. To think that we might be able to not just treat but cure all genetic-based diabetes with a single injection is incredible! The potential to save and change millions of lives is right at our fingertips. Although this is exciting at an individual level, it also helps solve issues at a societal level – in terms of time, resources, and cost. Solving long-term problems like these would be truly groundbreaking.
Analytical challenges
One downside of gene editing approaches is the risk of unwanted or unexpected changes at the protein level – so-called off-target effects. Ensuring the correct change in the genome does not automatically mean the results on the expression level are as expected. Cell networks are extremely complex and changing one variable can unexpectedly affect other parts or proteins. Developers must ensure their gene editing therapies do not introduce harmful off-target effects, which is why we need reliable and highly sensitive analytical methods.
Compared to traditional therapies based on small molecules, the analytical requirements for gene therapy applications are significantly more complex. Small molecules benefit from a very defined structure and purification processes built over decades. Newer therapies based on proteins often expressed in well-established cell lines already have increased complexity because they do not have a single defined structure, and the development processes and related analytical technologies are more sophisticated.
Gene therapies take this one step further. For instance, therapies or vaccines based on viral vectors consist of several proteins from the virus, contain the included gene, and are produced in a variety of different cell lines – which can impact impurities and post-translational modifications of the viral proteins. Production yields are also significantly lower for these types of therapies, which means that existing analytical setups need to be adapted. The latest analytical techniques detecting changes made to DNA or its transcribed products – the messenger RNA (mRNA) – are suitable even with minimal sample amounts. However, these techniques cannot determine the outcome on a protein-level: the phenotype. For instance, a desired gene modification could be successful on the gene level but also result in an unanticipated disturbance of protein interactions. Understanding the effects beyond genomics is a necessity for safe gene therapies.
The most common assays for protein-level verification involve immuno-based techniques, such as Western blot and ELISA, which require antibodies that may be difficult to obtain, of low quality, or too costly to produce at scale. And most importantly, how can we ensure that no other proteins were affected in case of a gene editing approach? Liquid chromatography coupled to mass spectrometry (LC-MS) is a critical key technology – its versatility and sensitivity enables product characterization and the ability to investigate changes, such as off-target effects.
Understanding gene editing by state-of-the-art proteomics
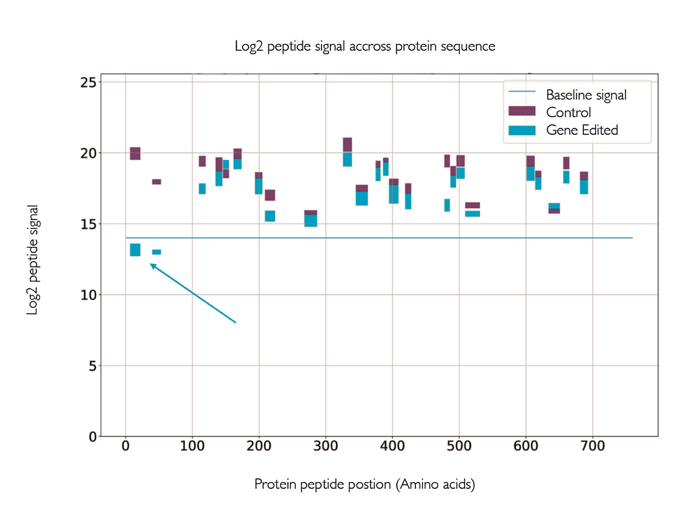
The latest developments in mass spectrometry allow us to tackle these challenges with confidence. Researchers are increasingly looking to understand and quantify gene editing induced changes across the entire proteome. The emergence of data independent acquisition (DIA) – an LC-MS/MS workflow that capitalizes on the speed and selectivity gains achieved with modern accurate mass spectrometry instrumentation – allows researchers to identify vast numbers of proteins more reproducibly than with conventional data-dependent acquisition (DDA) methods, while allowing for quantitative analysis from the same set of data (FIGURE 1).
Now, SCIEX has combined the speed and selectivity of DIA on the ZenoTOF 7600 system with significant sensitivity gains through the Zeno trap, which enables the identification of previously undetectable proteins with better protein sequence coverage, painting an even clearer and fuller picture of how gene editing affects other parts of the proteome. SWATH DIA provides the selectivity needed for such complex samples through its data-independent nature, ensuring accurate detection of as many relevant changes as possible. Partnering this technology with the enhancement of MS/MS spectral quality in terms of signal-to-noise results in Zeno SWATH DIA, which is key for detecting and quantifying the low-abundance changes in samples of low amounts.
Additional benefits for gene therapy applications
However, it does not stop there. Another key technology, unique to the ZenoTOF 7600 system, is electron activated dissociation (EAD). EAD is a newly developed dissociation approach that uses tunable electron energy, ranging from electron capture dissociation (ECD) to hot ECD and electron impact excitation of ions from organics (EIEIO), a mode comparable to electron impact (EI) dissociation. These modes can be used to produce varied fragmentation patterns for a wide range of modalities, including peptides. The technology offers more structural specificity compared with traditionally used collision-induced dissociation (CID) and can close gaps from existing electron-transfer dissociation (ETD) or ECD with chargestate independent efficiency.
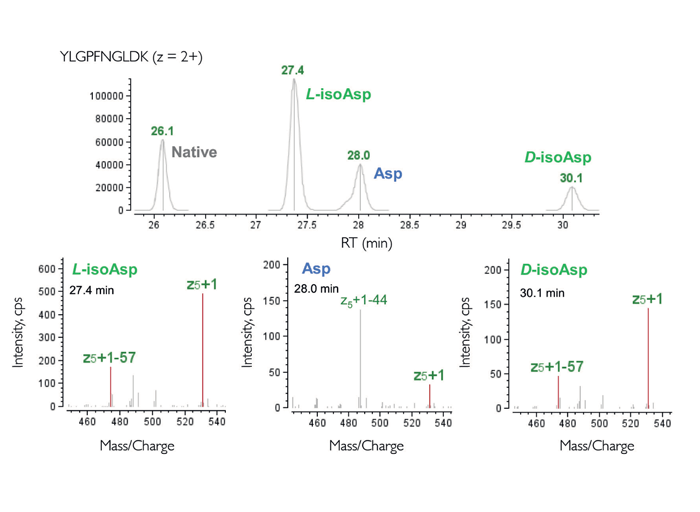
So, what does this mean for gene therapy application? This technology can enable single amino acid resolution for MS/MS spectral matches – something that CID struggles to achieve in the case of longer, difficult-to-fragment peptides and fragile modifications, such as glycosylations, phosphorylations, and sulfations. It can also identify isobaric amino acids, such as leucine versus isoleucine or aspartate versus iso-aspartate derived from deamidation through descriptive signature fragment ions (see figure 2).
Understanding peptide sequence and modifications, including their localizations, with certainty can greatly enhance biomarker ID and protein characterization work, which ultimately enables more streamlined development of new therapies. Liabilities, such as deamidations that could affect the uptake of viral vectors into the target cells for instance, can be understood and addressed early, removing guesswork from the equation.
Just as critical as obtaining the data for all analytical questions we seek to answer is the ability to process the raw data information in a streamlined, yet flexible manner. Although SCIEX provides access to the raw data, Biologics Explorer software is what allows us to really take full benefit from the extremely rich datasets. And that is vital! You can have the best MS system in the world producing the best data, but if the data cannot be converted into answers, it is essentially pointless. Having access to a capable software suite – the complete package – is very important.
What will the future bring?
Like any new field in medicine, drawbacks and failures are to be expected. We are still very much in a learning phase, as is the regulatory environment and the adoption of new technologies. And issues in clinical phases are usually linked to a lack of understanding of the therapeutic products. However, these modalities have the ability to truly make a difference – targeting what used to be undruggable targets and helping to cure diseases that were thought to be terminal. Very detailed characterization of these highly impactful medicines is absolutely necessary. Cutting corners in terms of product understanding is not an option; and with the ZenoTOF 7600 system offering Zeno SWATH DIA and Zeno EAD technology, we are well equipped to tackle analytical challenges. I am truly excited for the future of gene therapy and gene editing.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.




