The “-omic” era promises huge opportunities in personalized medicine. We should, however, remember that the real aim of this rapidly expanding field of research and analysis is to develop accurate, specific and cheap clinical tools that actually improve the treatment pathway. Translating discovery and validation efforts into clinical practice is challenging; nevertheless, it is possible, and we believe it is something that is being accelerated by the general introduction of mass spectrometry (MS) technology into clinical diagnostics.
Dispelling preconceptions
There’s long been a perception that immunoassay technology is superior to MS for routine diagnostics, not only with regard to sensitivity, speed, and cost, but also with regard to the perceived technical challenges associated with the latter, such as laborious sample preparation. But MS has advantages, and these are less often appreciated. In particular, it is much more specific than immunoassay; in fact, you may find a difference of three orders of magnitude between the MS-measured concentration of a given analyte and the immunoassay value. There are a lot of non-specific metabolite/protein immunoassays out there! This represents a potential problem for validation of MS-based protein quantitation where one approach is to assume that the molar concentration of a tryptic peptide represents the original protein concentration. Another, and we would argue more pragmatic approach, is simply to validate any peptide on the basis of intrinsic diagnostic specificity and sensitivity. In reality, it is the multiplexing potential of protein MS that provides the major advantage, obviously economic, over immunoassay by significantly reducing the cost per result.We have long believed in the potential of protein MS in clinical diagnostics; even so, we knew that if we were to get it more broadly used in clinical practice, we would need to simplify each step in the process – sample preparation, tryptic digestion, LC-MS/MS, and data analysis.
Where could MS prove useful?
First of all, we needed to identify potential clinical applications of protein MS. Based on our experience with MS/MS-MRM newborn dried blood spot (DBS) screening for inherited metabolic diseases, there was an obvious opportunity in newborn screening for sickle cell disease. It is worth remembering that, back in 1999, when we started our MS/MS-MRM work in this area, multiple reaction monitoring (MRM) techniques were not part of the toolkit; class compound scans, e.g. neutral loss for amino acids and parent ion for acylcarnitines, were seductive and had become the norm in newborn metabolite screening. This was despite the professional analytical view that quantitation of small molecules was best achieved using MRM analysis. More significantly, butylation of samples prior to MS/MS scanning was the accepted standard. Our first contribution to the routine use of MS/MS in newborn screening was to dramatically simplify the sample preparation process by demonstrating that the metabolites of interest could all be measured without butylation, i.e. non-derivatized metabolite screening. All that was required was simple elution from the DBS with methanol:water containing the appropriate stable isotopes, followed by flow injection. The second contribution was to directly target the metabolites of interest using MRM scanning. This represented two significant advantages. Firstly, inherited disorders could be targeted specifically, removing the ethical constraints imposed in the UK regarding class compound scans, i.e. the detection of disorders not sanctioned by the Newborn Screening Committee. Secondly, the precision of metabolite measurement was enhanced by a factor of 10 when compared with an equivalent data acquisition time for a class compound scan. The initial analytical validation was straightforward, but we needed to prove that underivatized MS/MS-MRM metabolite analysis was sufficiently robust for routine newborn DBS screening. To test this, we prepared 2,000 blood samples each from ourselves and a colleague, S Bird, and analyzed about 300 samples, including standards and controls, each working day for a month. The method proved to be exceptionally robust and overall coefficients of variation for metabolites, e.g. phenylalanine, were less than five percent. So we’d shown that MS/MS-MRM could be used in screening DBS for metabolites – and that presented us with a further huge opportunity. At that time, newborn hemoglobinopathy screening was just starting, using techniques that may appear archaic but are still in use in the majority of hematology laboratories today, i.e. isoelectric focusing (IEF) and HPLC. Both techniques can perform excellently in routine diagnostic laboratories but become challenging in the context of high-throughput newborn screening. However, from a protein MS perspective, newborn hemoglobinopathy screening appeared to be “low-hanging fruit,” because hemoglobin is a hugely abundant protein. Furthermore, we knew that if we developed something that worked, we had a market for it: about 750,000 newborns in the UK each year, all participating in an established screening program. Moreover, we didn’t need to invest in discovery work, because we already knew what the targets were; the genomics had already been done. It was time for us to put it to the test.Gathering the evidence
To begin with, we simply analyzed diluted whole blood by electrospray MS. This gives a beautiful charge series envelope, with the MS spectrum totally dominated by hemoglobin alpha and beta chains. Unfortunately, we considered the MS backgrounds to be too high and, because MS only reveals mass/charge (m/z) differences in the globin chains, not sufficiently mutation-specific. In addition, although sickle protein with a m/z shift of 30 daltons is relatively easy to detect, all the other mutations that combine with it to result in sickle cell disease differ by only 1 dalton in ~16,000. We could not differentiate these mutations on a standard triple quadrupole instrument. Our conclusion was that MS analysis was neither sufficiently sensitive nor specific for routine newborn DBS hemoglobinopathy screening. Our next step drew on our experience in the quantitation of small molecules. The idea was to employ tryptic digestion of blood followed by analysis of the resulting peptides in MRM mode. Trypsin, like other endopeptidases, preferentially acts at specific recognition sites in any protein, and therefore generates a consistent and reliable population of peptides – it liberates 15 peptides from human beta globin, for example. The brief, specified by the NHS Sickle and Thalassaemia Screening Programme, was to be able to detect hemoglobins S, C, DPunjab, OArab, and E. Earlier publications had demonstrated that the unique beta-chain peptides specific to these variant hemoglobins could be identified using chromatography and MS of tryptic digests of blood. This was reassuring, but we needed to demonstrate that we could detect each of the variant hemoglobins by MS/MS-MRM, using flow injection analysis, with sufficient sensitivity and specificity to provide a practical and robust solution for routine DBS newborn hemoglobinopathy screening. At least as important was the need to simplify traditional tryptic digestion procedures, which still are far too onerous to fit into any clinical process. Many laboratories complain if they have to add only one additional reagent to their existing process, so we knew we would have to keep the sample preparation simple.It was a revelation!
Solving these problems involved a lot of trial and error. With regard to sample preparation, initially we wanted to demonstrate the concept in whole blood, without any sample cleanup. It was also important to demonstrate that we could release the informative peptides, at least semi-quantitatively, within a one-hour trypsin incubation. We considered these to be the minimum criteria for a subsequent high-throughput clinical assay. In addition, if this were to be applied to newborn screening, flow injection would be an essential prerequisite. When it came to the actual mass spectrometry, we didn’t even know that we’d be better working with doubly charged rather than singly charged peptides. Our lack of understanding of peptide ionization and fragmentation, combined with an absence of peptide standards, meant that MS tuning using patient samples was challenging. Our initial full-range scans of whole blood tryptic digests looked horrible; there were plenty of signals, too many, and even when we zoomed in on the expected single- and double-charged m/zs expected for the wild-type beta chain T1 and sickle T1, the signals were not convincing and offered no prospect for formal tuning. Finally, in frustration, we programmed MS/MS product ion scans based on the theoretical single- and double-charged m/zs. It was a revelation! It was instantly obvious that doubly charged peptide fragmentation was much more informative. All of a sudden we could see a classic peptide spectrum, showing all the peptide signals as amino acids are lost from the N and C terminal ends.Progress was immediate: the most informative MRM for sickle peptide, i.e. adjacent to the mutation site, was identified, together with an equivalent MRM for wild-type peptide. Within an hour we demonstrated that, using rapid tryptic digests of whole blood, direct flow injection, and MRM mode analysis, we could differentiate normal wild-type beta T1 subjects, heterozygous sickle carriers, and homozygous sickle cell disease patients. (see Figure 1). That very same afternoon we were able to optimize the tuning parameters and set up the same procedure for each of the other sickling mutations. Subsequently, we collected a couple of hundred highly informative blood samples and analyzed them with our optimized method. We demonstrated 100 percent sensitivity and 100 percent specificity. The concept was patented by King’s College, London, and we eventually published the work in 2005 (1).
Did we answer our questions?
So we’d answered one of our primary questions: with minimal sample clean-up and a short tryptic digestion, the variant peptides can be detected sensitively and specifically using flow injection electrospray MS/MS with MRM data acquisition. But, had we done enough with regard to minimizing sample preparation and tryptic digestion in order for the assay to be acceptable in a routine newborn screening laboratory? Probably not! In fact, we addressed this by posing a new question: how would our assay perform in real-time neonatal screening? This required a real-world technical evaluation, and we were fortunate to receive funding for this from the NHS Sickle Cell and Thalassaemia Screening Programme. The evaluation was undertaken in collaboration with St James’ Hospital in Leeds, the idea being to compare our method with the IEF technology that they were using at the time.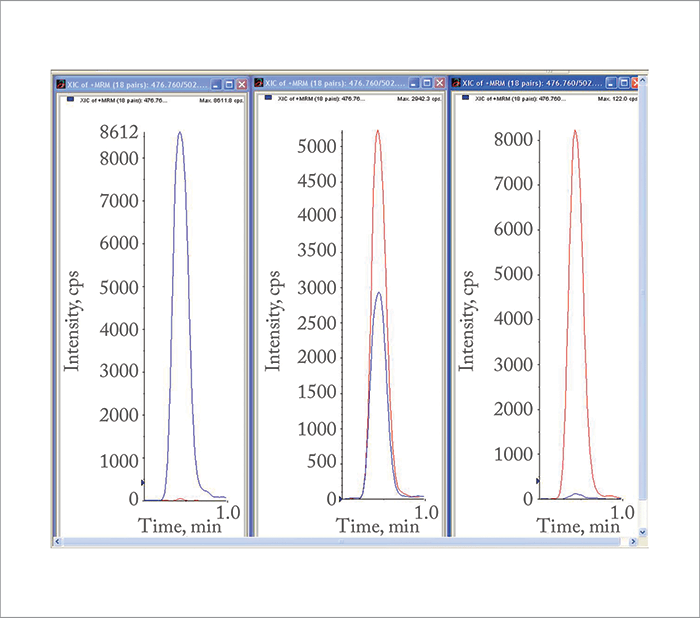
Duplicate punches were taken from neonatal DBSs in Leeds; one was analyzed by IEF, and the other was transported to us overnight, dry, in 96-well plates. When we received the samples we just added reagent, incubated for 30 minutes at 37° C, added stopping/dilution reagent, and injected 2 µL of sample using flow injection. The MS/MS-MRM analysis data acquisition time was 0.6 minutes. In effect, we had truly minimized sample work-up and data acquisition. The results were generated blind and were available within 24 hours, and they were excellent. From 40,000 neonatal DBSs, we identified all the sickling variants correctly, including 15 individuals with mutations associated with clinical disease. Just as important, in the context of newborn screening, there were no false negatives. It is worth emphasizing that in newborns, the beta chain is largely switched off, so as little as one percent of total hemoglobin may be beta chain. This means you need exquisite sensitivity, i.e. good signal to noise ratio, and the beauty of the MS/MS technique, in contrast to our original MS analysis, is the excellent signal to noise ratio.
We had validated the method – what next?
This was in 2008, and we now felt that we had validated our method, but how to get it into the routine newborn screening laboratory? We concluded that the easiest way forward was to develop a kit. We eventually produced a kit containing two reagents, one of which was a stable isotope internal standard, to provide a check, in every sample, on trypsin activity and instrument sensitivity. This was aimed at de-risking the process and ensuring that we would not miss any clinical cases. The stable isotope reagent also acts as a pre-analysis system suitability check of the MS/MS instrument. The concentration of the internal standard is set quite low so that if there are any instrument sensitivity problems, they can be identified before starting an analytical run. In summary, it’s a CE-marked kit, you only need two reagents, it takes only 30–45 minutes, it incorporates peptide standards for instrument setup, and it utilizes fully electronic data analysis with Chemoview on Sciex and NeoLynx on Micromass instrumentation, respectively.Application Longitudinal measurement of proteins in urine – for example, assessment of the albumin/creatinine ratio (ACR) to identify and monitor cardiovascular risk. Utility The urine ACR test can identify those at risk of cardiovascular disease 10–20 years before it becomes a clinical problem, and is increasingly claimed to be a highly useful early warning test for diabetic complications and general cardiovascular risk (5). Barriers Cost is perceived to be a barrier to implementation of urine ACR as a population screening tool, but may be manageable if we use a sample collection system similar to the DBS system.
At this stage, all we had to do was to convince potential clients to change to our kit. It is true, no one likes change! Although the NHS Sickle Cell and Thalassaemia Screening Programme had funded the successful study, it was only just over a year ago that the technique was officially designated as an acceptable test that could be purchased by NHS commissioners. Fortunately, in the interim, a far-sighted biochemist based in Cardiff, Stuart Moat, decided that the method was ideal for the Welsh Newborn Screening Programme. Stuart has now published NHS Wales’ experience with this kit (2) and, as a result, several other newborn screening laboratories in England have made the change. The kit is currently being used in Cardiff, Leeds, and Great Ormond Street Hospital, with significant interest from other laboratories in the UK, Europe, USA, Middle East, and India. We estimate that newborn DBS hemoglobinopathy screening, using MS/MS-MRM, will be performed on 250,000 babies next year in the UK.
Sixteen years of work; what next?
So yes, we’ve managed to get from basic science to the routine clinical laboratory – but remember it took about 16 years to do it! Nevertheless, we have shown that it is possible, and I expect to see other protein MS/MS-MRM based tests in routine clinical diagnostics in the next few years. A really important example, that has enormous potential, is measuring the protein content of urine – in particular the albumin/creatinine ratio (see box “MS/MS for Biomarker Detection in Urine”). This story started with the pioneering work of Harry Keen in the sixties, which showed that some patients with diabetes have increased protein excretion in their urine, specifically albumin (3). Importantly, Keen realized that the increased urine albumin was associated with the subsequent risk of developing diabetic nephropathy. More significant, but difficult to explain, was that increased urine albumin also identified the patients at risk of cardiovascular disease, 10–20 years before it became clinically evident. We now have large epidemiological studies, over 1.1 million people in the general population (~5 million subject years), which show that the risks of cardiovascular events are related to the urine protein (4). More recently, in a meta-analysis of 24 cohorts of about 600,000 people with no history of cardiovascular disease (median follow-up 4.2 to 19 years), the risks of cardiovascular mortality, coronary heart disease, stroke, and heart failure are all directly related to the urine albumin/creatinine ratio (ACR) (5). It really poses the question, as stated in the Lancet paper last year (5): when will clinicians appreciate the importance of ACR, as a very early warning test, in determining diabetic complications and general cardiovascular risk? Some clinicians are skeptical because of the relatively high biological variability of ACR tests, but this can be addressed with a multiple sampling approach: we always take the geometric mean of three early morning urines, which smooths out the natural variation. Transportation and storage of urine samples is costly, but the sensitivity of MS/MS-MRM means that urine samples can be collected on filter paper – just as blood is collected for newborn screening – or other micro-sampling devices. This raises the possibility of developing personalized medicine, where individuals take control of monitoring their own health by collecting micro blood and/or urine samples at home and posting to a referral laboratory. If this is done regularly, e.g. annually, within a few years, the intrinsic individual variation of any diagnostic test can be assessed and a within-individual normal range established. The result is very early warning of developing disease including diabetes, cardiovascular, renal, liver, and many cancers. Early detection means that lifestyle changes can be made or appropriate therapies started, with the potential to reverse, stop, or ameliorate disease progression. In conclusion, the introduction of routine clinical protein MS has taken many years, but in the future, now that the MS hardware is more established, the research bench to routine clinical diagnostic timeline should be shorter. We believe that tryptic peptide analysis by electrospray MS/MS-MRM has significant clinical utility and will play a key role in both population screening and personalized medicine: sample collection is easy, the patient pathway can be optimized, and it can be made cost-effective by simplifying the analytical logistics and exploiting the multiplexing capability of MS. Neil Dalton is Professor of Pediatric Biochemistry, King’s College London; Director of the WellChild Laboratory, Evelina London Children’s Hospital and SpOtOn Clinical Diagnostics Ltd. Charles Turner is Deputy Director of the WellChild Laboratory, Evelina London Children’s Hospital and a Director of SpOtOn Clinical Diagnostics Ltd.First published in The Pathologist (www.thepathologist.com), a sister publication of The Analytical Scientist.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- YA Daniel et al., “Rapid and specific detection of clinically significant haemoglobinopathies using electrospray mass spectrometry-mass spectrometry”, Br J Haematol, 130, 635–643 (2005). PMID: 16098080. SJ Moat et al., “Newborn blood spot screening for sickle cell disease by using tandem mass spectrometry: implementation of a protocol to identify only the disease states of sickle cell disease”, Clin Chem, 60, 373–380 (2014). PMID: 24158758. H Keen et al., “The concomitants of raised blood sugar: studies in newly-detected hyperglycaemics”, Guys Hospital Reports 118, 247–254 (1969). PMID: 5804067. K Matsushita et al., “Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis”, Lancet, 375, 2073-2081 (2010). PMID: 20483451. K Matsushita et al., “Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: a collaborative meta-analysis of individual participant data”, Lancet Diabetes Endocrinol, 3, 514–525 (2015). PMID: 26028594.



