The problem
The problem originates from the pharmaceutical industry: the need to unambiguously identify trace impurities that nearly co-elute with the main active pharmaceutical ingredient (API). The identification can be easily achieved by nuclear magnetic resonance (NMR) spectroscopy, but it requires that about 1 mg of the impurity be prepared at a purity level of at least 90 percent. Given the current state of instrumentation, the pharma industry could not find acceptable technologies capable of producing a (nearly) co-eluting trace impurity at this level within a reasonable timeframe – either because of a lack of resolution power (semi-prep instruments, see Figure 1) or a low production rate (analytical instruments). An alternative semi-prep technology is therefore desperately needed to solve this problem.
Background
First of all, by “recycling” liquid chromatography, we do not mean some sort of “green” separation process based on a clean or regenerated mobile phase. The main challenge that we attempted to tackle with this technique is the need for unprecedented high-resolution performance – resolution that cannot be realized even with today’s state of the art ultra-high pressure LC systems (up to 1.5 kbar) and columns packed with very fine particles (< 2 µm).
We envisage our technique primarily being applied when compounds have very similar structures (take isomers or isotopes for example) and cannot be baseline separated, even after intense screening to optimize mobile and stationary phases or with the use of 2D-LC systems.
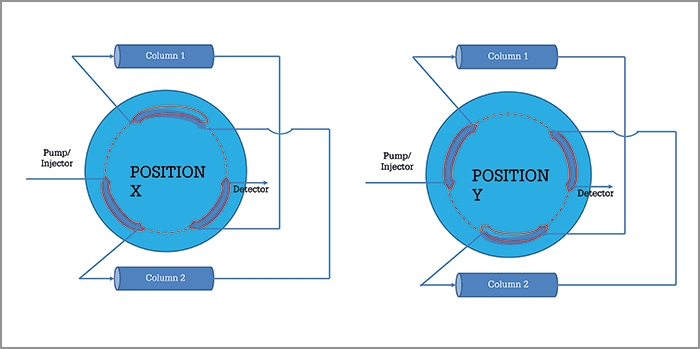
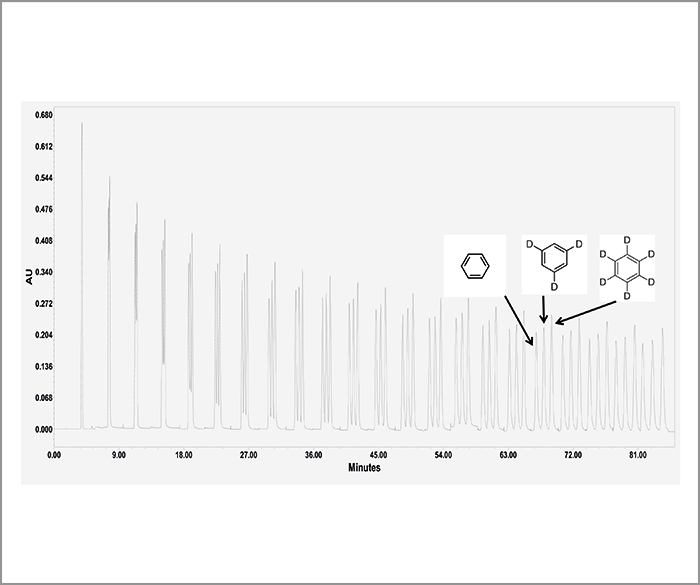
The core concept behind recycling chromatography is quite straightforward, and was first proposed in preparative LC in the 1960s, when particle sizes were too large (> 30 µm) and the resolution power was too low. The use of a two-position valve and a pair of twin columns, results in a virtual infinitely long column that can still be operated at low pressures with standard pumps. The key part of the recycling technique consists in re-injecting the targeted mixed sample zone from the outlet of one column into the inlet of the second twin column (see principle in Figure 2). Twin column recycling chromatography was then born (1) and there are many successful applications of this technique under analytical conditions ([2]; see one example in Figure 3). We then designed a new semi-preparative purification technique (high-resolution semi-preparative liquid chromatography) including the above-mentioned recycling LC unit.
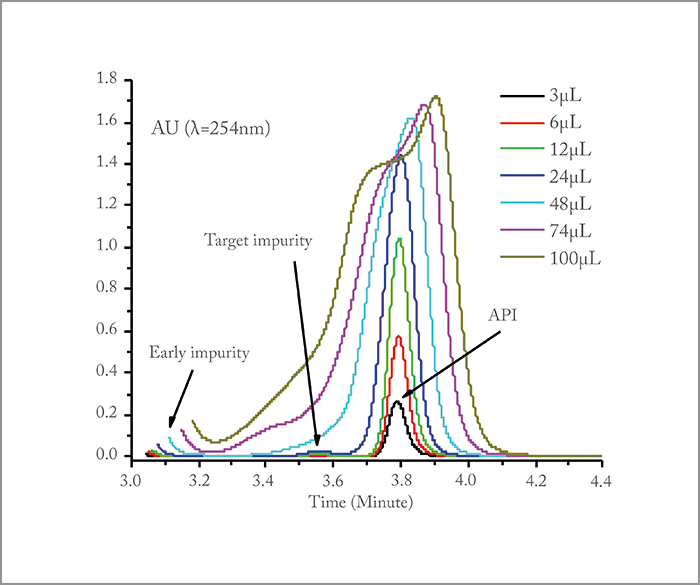
The problem of detecting trace impurities in pharmaceutical manufacturing requires an unusual combination of system capabilities: not only ultra-high performance levels (to isolate the impurity from the API) but also a separation system with semi-preparative capabilities (to produce 1 mg of material). High-resolution chromatography can be achieved today with classical HPLC, UHPLC, or 2D-LC heart-cutting systems. Yet, these systems operate under linear chromatography at very small injection volumes and low impurity/API concentrations. It would take years before 1 mg of isolated impurity could be prepared – not an acceptable solution. On the other hand, currently available preparative instruments such as batch Prep-LC, simulated moving bed (SMB), or steady state recycling (SSR) systems can easily prepare 1 mg of material in a very short time. However, they cannot cope simultaneously with a very low selectivity factor between impurity and API, an extremely low relative abundance of the impurity relative to the API (~ 1/1000), and a severe mismatch between the properties (elution strength, viscosity) of the sample diluent and those of the eluent, causing severe band distortion (see Figure 1). We were therefore asked to design a new separation system capable of combining incompatible analytical (small sample volumes and concentrations) and preparative (large sample volumes and concentrations) features. In brief, a real conundrum stood in front of us.
The solution
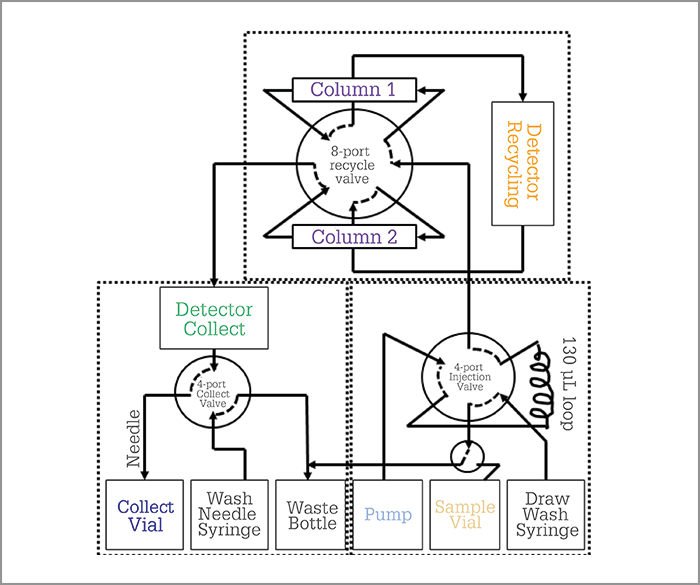
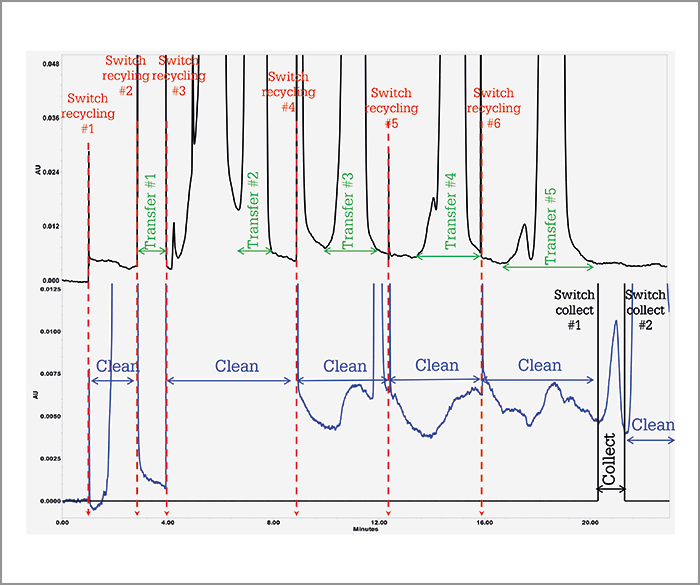
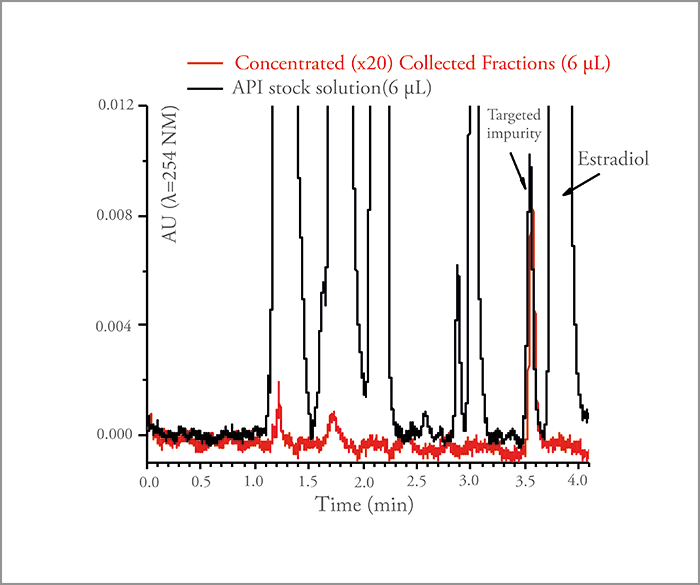
To solve the pharma industry’s problem, we had to build an integrated system capable of injecting large volumes of very concentrated API samples (~ 10 g/L), separating the two distorted bands (impurity and API) by performing recycling chromatography, and collecting the isolated impurity using a fraction manager – the high-resolution semi-preparative LC (HR-SP-LC) system (Figure 4). The two most critical features of the system are i) the temperature control of the pair of twin columns for the sake of cycle-to-cycle and injection-to-injection repeatability and ii) the minimization of sample dispersion upon sample transfer from the first to the second column. Figure 5 demonstrates the success of the HR-SP-LC system in the case of the isolation of an unknown impurity present on the left side of the main API (10 g/L estradiol sample). It required a total of six cycles to achieve a complete baseline separation with an enrichment factor of the impurity relative to the API in the order of 5000 (Figure 6).
Of course, this principle was tried nearly 50 years ago in preparative recycling chromatography, but for much less challenging separation problems: the goal was essentially to prepare pure fractions of the most abundant compound present in the sample mixtures by GC (no problem of diluent-to-eluent mismatch) or by LC (relatively easy purification at moderate or low yield). In the present case, the targeted impurity is present in trace amounts at concentrations as low as a few mg/L while the concentration of the nearly co-eluting API compound is as large as tens of g/L. Therefore, the yield must remain close to 100 percent, as we cannot afford to lose any of the targeted impurity; meanwhile, we have to maintain a high purity level (> 90 percent for NMR characterization).
Additionally, the performance of today’s pumps (high pressure) and valves (low dispersion valve), as well as the quality of connections (face seal connections) between the different parts of the HR-SP-LC system have improved so much that it is now possible to use modern UHPLC columns packed with very fine particles without losing too much resolution upon transferring the analyte from the first to the second column. This was not possible decades ago. Finally, the stability of the column temperature set by today’s active eluent preheaters ensures an unprecedented reproducibility of the chromatograms shown in Figure 5 and a complete automation of the system including sample injection, recycling separation, and fraction collection. The RSD of the retention times remains within 0.3 percent over an entire week (5). The process can be kept running for weeks without user intervention and still provide the targeted purity level for each of the collected fractions.
In principle, the experimental set-up (Figure 4) is not more complicated than that of a standard 2D-LC process, incorporating: a sample manager that can accommodate large sample volumes (50 mL vials), a two-position six-port valve, a two-column oven with active preheater, a fraction manager connected to the recycling unit, one low-dispersion detection cell, and one standard chromatography software to control the quality of the purification process immediately before collection of the trace impurity. Once these different units are connected to each other and commanded by the same chromatographic software, the HR-SP-LC system can be entirely automated by tabulating the times at which the recycling and collection valve should be switched. Therefore, a non-specialist analytical chemist can easily run the process. The only experimental information requested from the user is the measurement of the retention time of the targeted impurity after elution through one column length. Once this retention data is known, the user defines a unique isocratic chromatographic method (sample injection volume, flow rate, temperature, eluent composition, impurity band shaving times, recycling and collection times), the method is validated (or not) once by the signal recorded immediately before the fraction collector, and a sequence of as many runs as necessary to collect the desired amount (1 mg) of pure (>90 percent) impurity is programmed.
Beyond the solution
As yet, the system is still a prototype. We are still experiencing challenges in terms of production rate; so far, the optimum production rate is around a few micrograms per hour using standard 4.6 mm i.d. x 150 mm long analytical columns and injection volumes up to 250 µL. If the initial impurity concentration is only 1 mg/L, one can only produce about 0.1 mg of impurity per week for a half an hour run and six cycles. This is clearly insufficient (1 mg is needed within a day). Therefore, we are planning on scaling-up the HR-SP-LC process by using 10 mm i.d. × 250 mm long columns packed with 5 µm particles. Each injection will consist of a 1–2 mL sample injection volume. This will bring the production rate up to the desired level, but will add three new challenges: i) how to accurately control the temperature of these large i.d. columns with a wide air-convection oven, ii) how to avoid the undesirable band distortion caused by enhanced viscous fingering and diluent-to-eluent mismatch effects, and iii) how to minimize the increased inter-column dispersion when using wider connecting tubes (i.d. × 2) and higher flow rates (×5). Scaling-up the HR-SP-LC process will likely mean applying larger numbers of cycles to achieve the same purity level – but, overall, production rates are still expected to increase.
We have focused specifically on pharmaceutical applications, but there are other areas where this approach could bring significant value. The problem we have tackled here is a very general one and is frequently encountered in other separation fields: trace impurities are everywhere and they need to be unambiguously identified for safety/quality controls. One interesting application besides pharma applications is the unambiguous identification of aggregated forms of monoclonal antibodies (mAb), which nearly co-elute with their monomeric form and require complimentary spectroscopic techniques beside conventional separation techniques. This can be done by recycling size exclusion chromatography (2). In addition, the plastics industry would benefit greatly from analyzing the composition of their synthesized polymers by coupling a HR-SP-LC system with NMR spectroscopy. Indeed, for any application where the separation and preparation of a sufficiently large amount of pure impurity remains the limiting step, HR-SP-LC could be of great benefit.
Fabrice Gritti is a Principal Research Scientist at Waters Corporation, Milford, Massachusetts, USA.
Acknowledgements
The author would like to thank Mark Basile, Sylvain Cormier, Dennis DellaRovere, Michael Fogwill, Martin Gilar, Thomas McDonald (Waters, Milford, MA, USA), Frank Riley and Qi Yan (Pfizer, Groton, CT, USA) for their constant technical and material supports to this research project.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- J Porath, H Bennish, Arch Biochem Biophys, 1, 152–156 (1962). F Gritti et al., J Chromatogr A, 1524, 108–120 (2017). F Gritti, S Cormier, J Chromatogr A, 1532, 74–88 (2018). K Bombaugh, R Levangie, Separation Sci, 5, 751–763 (1970). F Gritti et al., J Chromatogr A, 1566, 64–78 (2018).



