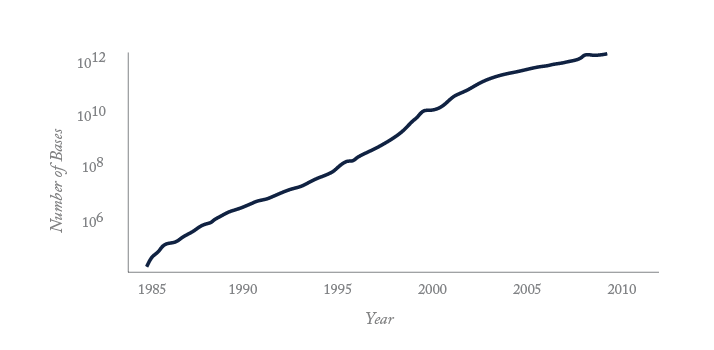
Progress in genomics, powered by advances in analytical chemistry, is transforming the study of life from a qualitative to a quantitative science. The continuous introduction of new approaches has enabled speed and sequencing capacity to rocket while costs have plummeted (see “Three Decades of Growth”). It cost several billion dollars to sequence the first human genome. Today, twelve years later, entire genomes are being sequenced for $10,000 US dollars. Undoubtedly, the pace will accelerate still further as analytical chemists and engineers develop faster and cheaper sequencing technologies. This revolution will likely require generations of medical scientists to interpret, but ultimately will deliver major advances in health care.
Here, we look back at the origins of DNA sequencing, describe the range of approaches that are in use today and sketch the technologies that will drive the next phase of genome sequencing.
The field of genomics began with the discovery of DNA structure by Watson and Crick in 1953 (1). That structural model has served as a template for our understanding of the function of the molecule, although details remained elusive for two decades until the development of two powerful methods for the determination of nucleotide sequences in DNA. Initially, the chemical degradation method of Maxam and Gilbert was the tool of choice (2). However, that reaction used rather hazardous chemicals, and was supplanted in the late 70’s by Sanger’s dideoxy chain termination method, once the necessary reagents became commercially available (3). Sanger’s method employs biochemical synthesis; a primer (which provides a starting point for DNA synthesis) is annealed to one strand of DNA in the presence of DNA polymerase, deoxynucleotide triphosphates, and a small amount of a single dideoxynucleotide. The DNA polymerase adds deoxynucleotides to the primer, synthesizing the complementary strand of DNA. Occasionally, a dideoxynucleotide is incorporated, which produces a strand that cannot be further extended; this addition is called a chain-termination reaction. Four separate reactions can be performed with different dideoxynucleotides. The reaction products are then separated in different lanes in a polyacrylamide gel. Sanger employed radioactive nucleotides and detected the fragments by autoradiography. Autoradiography-based sequencing throughput is ultimately limited by the visual interpretation of sequencing gels, which is a tedious and error-prone step in the procedure. The next giant leap in sequencing technology came a decade later with the development of fluorescence-based automated sequencers. Leroy Hood’s group at Cal Tech (4) and James Prober’s group at DuPont (5) extended Sanger’s method by replacing the radioactive label with fluorescent tags, and replacing autoradiography with laser-induced fluorescence detection.
In Hood’s technology, a primer is labeled with one of four fluorescent tags. Each dye-labeled primer is used in a chain-termination reaction with a single dideoxynucleotide. For example, a primer labeled with a red dye might be used with the dideoxyadenosine chain terminating reaction, a green dye used with the dideoxycytidine reaction, a yellow dye with the dideoxyguanosine reaction, and a blue dye with the dideoxythymidine reaction. The reactions products are pooled and separated in a single lane of a polyacrylamide gel. A four-color spectrograph records the fluorescence intensity as fragments migrate past the fixed detector. The identity of the terminating nucleotide is determined based on the spectral characteristics of the dye. Prober developed a more sophisticated version of this technology where the chain terminating reaction is performed simultaneously with the four labeled dideoxynucleotides, simplifying the overall reaction. Hood’s and Prober’s technologies replaced the laborious and error-prone steps of visual interpretation of an autoradiogram generated from a sequencing gel. And it was this automated reading of DNA sequence that catalyzed the human genome project. In 1986, a group of visionary biochemists realized that this new analytical technology could be used for the very large and ambitious goal of determining the sequence of the entire human genome. Initial efforts were funded first by the Department of Energy and then the National Institutes of Health in the U.S. Gradually, it became an international effort, drawing on scientific and financial contributions from many countries.
Despite the success of fluorescence-based sequencing, the systems were not fully automated. Preparation of the sequencing gels remained extremely laborious, particularly when envisioning such an ambitious program, so a modest portion of the genome project funded improvements in the analytical technology. A number of approaches were investigated, including mass spectrometry, scanning probe microscopy, and microfabricated technologies. Unfortunately, those approaches lacked sufficient sensitivity, speed, robustness, or resolution and were abandoned. Ultimately, technology based on capillary electrophoresis was developed by academic and industrial researchers, and successfully commercialized by Applied Biosystems and Amersham. The flexibility and robustness of fused silica capillaries facilitated the automation of the electrophoretic separation, in addition to the reading of the sequence using laser-induced fluorescence. The Applied Biosystems instrument, the model 3700 DNA sequencer, introduced in 1998, was the most successful and generated the lion’s share of the sequence data for the first human genomes. That instrument was based on technology developed by Hitachi in Japan (6) and the University of Alberta in Canada (7). In an interesting business decision, Applied Biosystems created a new company, Celera, whose goal was to use whole-genome shotgun sequencing, wherein a huge number of relatively short fragments were sequenced using ABI’s model 3700 instrument. The overlap of these shotgun sequences was used to assemble the data into a coherent sequence. This technology had been pioneered by J. Craig Venter in earlier efforts to sequence smaller genomes. In contrast, the publically funded effort employed a series of mapping and cloning steps using a range of vectors in a more ordered approach. The commercial and publically funded projects proceeded in parallel, and they ended in a dead heat, describing their results in a set of publications that appeared in Science and Nature in February 2001. Over a billion dollars was invested in the sequencing effort to determine the three billion bases of sequence in the human genome, which was completed well ahead of schedule and well under budget. The use of extensive mapping and cloning of a large genome has not been replicated. Instead, all modern sequencing efforts employ some form of whole-genome shotgun sequencing.
Capillary electrophoresis remains a useful tool for routine sequencing of small fragments. However, a suite of new technologies has been developed, providing spectacular improvements in analytical performance. Four such technologies are compared in Table 1.
KEY: SE: single-end sequencing, PE: paired-end sequencing, bp: base pairs, gb: gigabase, tb: terabase.
Data taken from Liu et al. “Comparison of Next-Generation Sequencing Systems” J. Biomedicine & Biotechnol. 2012, Article ID 251364
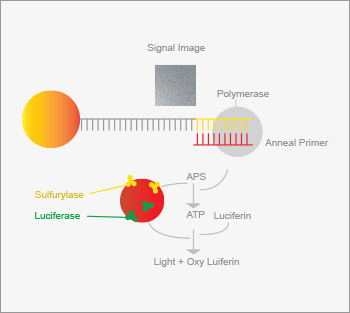
A group at the Royal Institute of Technology in Stockholm developed an alternative to Sanger sequencing (8). The fragment to be sequenced is immobilized on a solid support along with a primer. DNA polymerase and a single deoxynucleotide triphosphate are then added. If the deoxynucleotide is incorporated into the strand, a diphosphate group (pyrophosphate) is produced. This molecule is converted to ATP by sulfurylase. The ATP acts as a substrate for luciferase, generating a flash of light upon incorporation of the nucleotide. If the nucleotide is not incorporated, no light is observed. The DNA sequence is then determined by treating the sample with different nucleotide triphosphates in series, and recording which nucleotide generates luminescence. The sequence of several hundred nucleotides can be determined in a single run. Pyrosequencing forms the basis of one of the earliest next-generation sequencing methods, which was developed by 454 Life Sciences, a subsidiary of Roche (9). It is a massively parallel form of pyrosequencing where millions of templates are sequenced simultaneously. Genomic DNA is fragmented and bound to beads under sufficiently dilute conditions to ensure that no more than one fragment is attached to a bead (see Figure 1). Beads are compartmentalized in an oil emulsion and the polymerase chain reaction (PCR) is used to amplify the fragments, creating a coat of 107 copies of the original sequence on the bead. Beads containing amplified fragments are enriched and captured into wells on a slide. Appropriate enzymes required for pyrosequencing are added to the wells and each of the four deoxynucleotides is pumped in succession over the slide and luminescence recorded. Although each step is relatively slow, sequencing data is generated from tens of millions of samples in parallel producing a gigabase of sequence per run, which is six orders of magnitude higher throughput than that produced by capillary electrophoresis instruments.
A group at the Royal Institute of Technology in Stockholm developed an alternative to Sanger sequencing (8). The fragment to be sequenced is immobilized on a solid support along with a primer. DNA polymerase and a single deoxynucleotide triphosphate are then added. If the deoxynucleotide is incorporated into the strand, a diphosphate group (pyrophosphate) is produced. This molecule is converted to ATP by sulfurylase. The ATP acts as a substrate for luciferase, generating a flash of light upon incorporation of the nucleotide. If the nucleotide is not incorporated, no light is observed. The DNA sequence is then determined by treating the sample with different nucleotide triphosphates in series, and recording which nucleotide generates luminescence. The sequence of several hundred nucleotides can be determined in a single run.
Pyrosequencing forms the basis of one of the earliest next-generation sequencing methods, which was developed by 454 Life Sciences, a subsidiary of Roche (9). It is a massively parallel form of pyrosequencing where millions of templates are sequenced simultaneously. Genomic DNA is fragmented and bound to beads under sufficiently dilute conditions to ensure that no more than one fragment is attached to a bead (see Figure 1). Beads are compartmentalized in an oil emulsion and the polymerase chain reaction (PCR) is used to amplify the fragments, creating a coat of 107 copies of the original sequence on the bead. Beads containing amplified fragments are enriched and captured into wells on a slide. Appropriate enzymes required for pyrosequencing are added to the wells and each of the four deoxynucleotides is pumped in succession over the slide and luminescence recorded. Although each step is relatively slow, sequencing data is generated from tens of millions of samples in parallel producing a gigabase of sequence per run, which is six orders of magnitude higher throughput than that produced by capillary electrophoresis instruments.
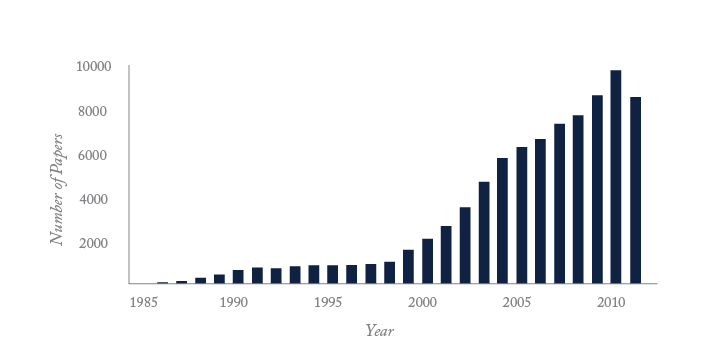
Only consumer electronics and computation have undergone the rapid technological growth seen in genomics and proteomics. That growth is illustrated by the number of publications indexed by Medline and the growth in the size of Genbank, the public repository for genomic sequence. Bases deposited doubles every 18 months, an extreme example of Moore’s law, a computing term from the1970’s which held that processing power for computers would double every two years. And the Genbank data are the tip of the iceberg; much more sequencing data reside in private databases. The journal Nature reported in July 2012 that 10,000 human genomes have been sequenced, which corresponds to 3 x 1013 bases of finished sequence. When considering the redundancy with which genomic sequence is obtained, it is likely that well over 1015 bases of genomic sequence have been generated, the vast majority in the past 12 months.
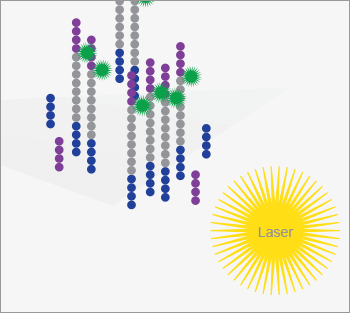
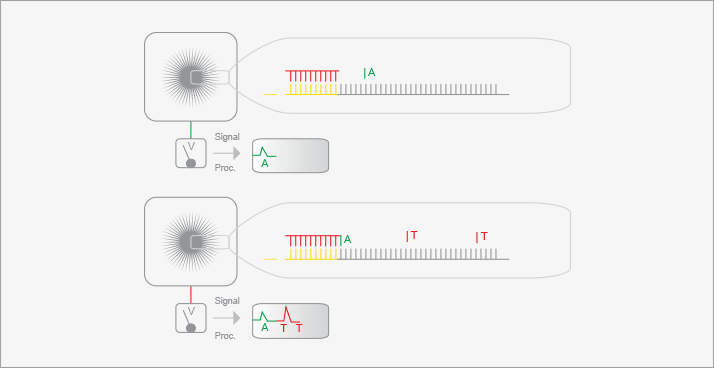
Illumina offers another next-generation sequencing method (10). Sequencing templates are immobilized on a surface to generate a “lawn of sequencing primers” (see Figure 2). A clever surface-based version of PCR is used to amplify each of the sequencing fragments and to immobilize those fragments in a micrometer region on the surface, generating clonal clusters. Sequencing is performed by hybridizing a primer to the immobilized fragments, with the addition of DNA polymerase and a modified deoxynucleotide triphosphate. This modified nucleotide contains a fluorescent tag that allows detection of the nucleotide once it has been incorporated onto the growing complementary strand. It also has a blocking group that prevents addition of another nucleotide. Excess labeled nucleotides are washed from the system, and a laser-based fluorescence detector images the surface. Those templates that incorporated the modified nucleotide are detected based on their fluorescence. The label and the blocking group are then removed chemically, and the process is repeated with another nucleotide, building up the sequence of each of the immobilized templates. This technology produces huge amounts of data, ~5 x 1010 bases per run, but each template only generates ~100 bases of sequence, which can challenge the assembly of the individual sequences into a complete genomic sequence; the instrument is ideal for resequencing known genomes rather than de novo sequencing of novel organisms. Because of this massive data output, Illumina currently controls around two-thirds of the sequencing market share.
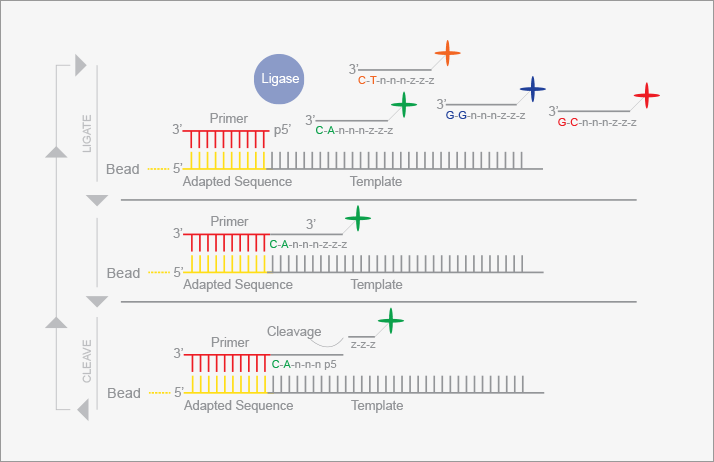
SOLiD (Sequencing by Oligonucleotide Ligation and Detection, see Figure 3) also generates short sequences from large numbers of templates (11). Like the 454 technology, templates are amplified and immobilized on a solid particle based on emulsion PCR. Tens to hundreds of millions of beads are deposited on a glass slide and the sequence read by addition of a fluorescently labeled oligonucleotide. A ligase is used to covalently attach the oligonucleotide to the primer. Only those oligonucleotides whose sequence matches the complementary portion of the template are attached. The slide is imaged by laser-induced fluorescence, and clever chemistry is used to remove the fluorescence dye. The procedure is repeated, gradually extending the complementary strand. This procedure interrogates two nucleotide positions at a time, examining the 4th and 5th nucleotide in the template. Once the procedure is completed, the synthesized strand is removed, and a new primer is added which has an additional base, allowing the determination of bases that were missed in the first pass. A set of five primers is used to determine the sequence across ~25 to ~50 nucleotides. The instrument is capable of generating over 2 x 1010 bases of sequence per day, and of sequencing a genome in a few days.
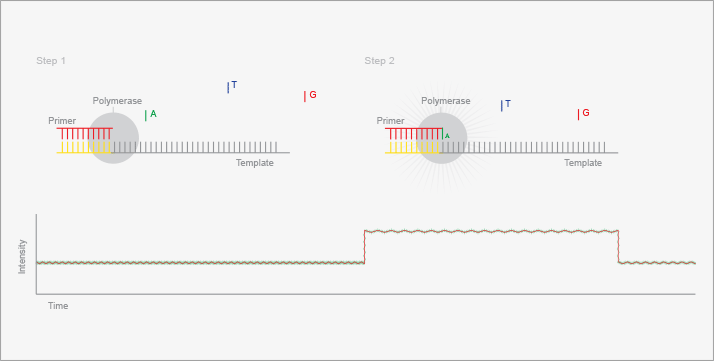
Ion Torrent (see Figure 4) employs similar chemistry to pyrosequencing and relies on the scalable manufacturing pipeline developed by the semiconductor industry (12). Unlike the other detection schemes, however, Ion Torrent utilizes non-photonic detection. The addition of a deoxynucleotide triphosphate to a growing oligonucleotide results in the formation of a proton in addition to the pyrophosphate group. This proton causes a drop in the pH of the solution, which can be detected with a high sensitivity pH meter. Ion Torrent deposits pH sensors at the bottom of a huge number of wells on a microfabricated device. Sequencing proceeds by amplifying large numbers of DNA fragments, depositing them into the wells, and then flowing DNA polymerase and a deoxytriphosphate over the device. Those wells that contain a fragment that incorporated the nucleotide will experience a pH decrease, which is sensed by the electronics. Even though only 20-40 percent of the wells are used in an Ion Torrent chip, the massively parallel nature of the design still results in high data output. The manufacturer claims that over 4 x 1011 bases of sequence are generated in a two-hour operation, with over 99.5 percent accuracy.
There has been significant interest in extending whole genome sequencing to the general population as a guide to individualized medicine. The National Human Genome Research Institute (NHGRI) has invested heavily in translating the genomic sequence into guidance for the treatment of patients at their bedside. However, for individual genomic sequencing to be a reality, costs must drop by one to two orders of magnitude. To this end, NHGRI has active funding solicitations with the goal of obtaining whole genome sequences for $1000. In 2008, Helicos became the first company to offer what it termed “True Single Molecule Sequencing” with its HeliScope technology (13). A short DNA strand (25 bp) is prepared and a PolyA primer added to the 3’ end. Up to a billion DNA strands are hybridized to a DNA flow cell with Oligo dT capture sites. Sequencing reactions are detected from individual molecules with the addition of fluorescent-labeled nucleotides. In its marketing, Helicos placed great emphasis on freedom from amplification, citing errors in the PCR process as a major flaw with other sequencing approaches. Unfortunately, Helicos was unable to generate sufficient cash flow to service their debt, and they filed for Chapter 11 bankruptcy on November 15, 2012. Pacific Biosciences has also developed a single molecule detection approach, termed SMRT for Single Molecule Real Time sequencing (14). SMRT and PacBio, as it is commonly called, initially generated a lot of buzz in the genomics world. The SMRT system (see Figure 5) offers unlimited read length, no need for PCR amplification, and fast detection. The methodology is based on using DNA polymerase as a real-time sequencing engine. Multiple DNA polymerases are immobilized in Zero Mode Waveguides and then monitored via fluorescence for the addition of labeled nucleotides. The approach is very promising, but has not been readily adopted by the genomics community because of the expensive instrumentation, coupled with a higher relative error rate compared to other sequencing approaches.
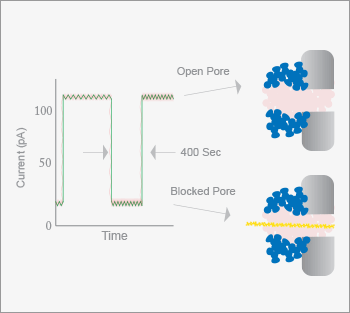
In February, 2012, Oxford Nanopore announced the development of their sequencing platform, based on passing single DNA strands through protein nanopores embedded in a polymer membrane (see Figure 6). As the nucleotides pass through the protein channel, a detectable change in the membrane current occurs (15). Each of the four nucleotides produces a distinct signal, allowing discrimination and ultimately, a DNA sequence. If a hairpin structure is present, both the sense and antisense strands can be read for each DNA molecule. The approach can also be performed in an “exonuclease-sequencing” fashion, where an exonuclease is positioned over the nanopore and the individual nucleotides are cleaved and translocated through the pore one at a time. Nanopore is currently generating interest for several reasons. The nanopore technology does not appear to have read length limitations. During the initial press conference, Oxford Nanopore announced that they had sequenced a 48 kb virus genome in a single pass. Another appealing aspect is cost: Users do not need to purchase expensive instruments, buying only a disposable sequencer, which is about the size of a USB memory stick, for an estimated $900. At the time of writing, the Nanopore system was not available for sale nor has a peer-reviewed article demonstrating the capabilities of the technology been published – two events that are eagerly anticipated by the genomics community.
Nearly three decades of technology advances have now reduced the cost and speed of genomic sequence to the point where whole genome studies are routine. Further advances are on the horizon, but we are approaching the point of diminishing returns; each individual genome is generally static, so there is little need to resequence. The obvious exception is in tracking cancer progression, where loss of tumor suppressor genes carries a grim prognosis. Genomic analysis is likely to reach maturity within a decade, when the cost of sequencing a human’s genome drops to ~$100. We have not addressed epi-genomics, which deals with post-replication modification of specific nucleotides in the genome. Most common is the incorporation of a methyl group in cytosine residues, which has implications in gene expression. In comparison to genomics, epi-genomics is in its infancy. There is a real need for improved technologies to produce low cost, high-speed analysis of the epi-genome, which will be invaluable in understanding developmental biology and disease progression. We have also not addressed transcription analysis. The use of reverse transcription and either hybridization arrays or whole-genome sequencing have become powerful tools in transcriptomics. Real-time PCR provides quantitation across many orders of magnitude. Technology for transcriptomics is likely to reach maturity within a decade of the maturation of genomics.
Freeman Dyson said “New directions in science are launched by new tools much more often than by new concepts” (16). The astonishing advances in genomics over the past two decades are a striking demonstration of this statement. Within living memory, the determination of a few bases of DNA sequence would require a graduate student's entire career. Today, whole genome sequences are generated within a few days at core facilities located on many university campuses. This advance in technology results from a set of wise choices first by the Department of Energy, then by the National Institutes of Health, and now by the private sector. First, these funders identified a problem and articulated it clearly. Second, they provided significant funding levels for a wide range of technologies. Third, they culled unsuccessful technologies. Fourth, they nurtured those technologies that showed progress toward the solution of the problem. This model has proven to be remarkably robust in the development of analytical instrumentation, and has provided an astonishing return on investment to the public and private sectors.
Norman J. Dovichi and Amanda B. Hummon are at the Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- J. D. Watson and, “Molecular structure of nucleic acids; a structure for deoxyribosenucleic acid,” Nature, 171: 737-8 (1953). A. M. Maxam and W. Gilbert, “A new method for sequencing,” Proc. Natl. Acad. Sci. USA, 74: 560-4 (1977). F. Sanger, S. Nicklen and A.R. Coulson, “DNA sequencing with chain-terminating inhibitors,” Proc. Natl. Acad. Sci. USA, 74: 5463-7 (1977). L. M. Smith, et al., “Fluorescence detection in automated DNA sequence analysis,” Nature, 321: 674-9 (1986). J. M. Prober, et al., “A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides,” Science, 238: 336-41 (1987). H. Kambara and S Takahashi, “Multiple-sheathflow capillary array DNA analyser,” Nature, 361: 565-6 (1993). D. Y. Chen, et al., “Low-cost, high-sensitivity laser-induced fluorescence detection for DNA sequencing by capillary gel electrophoresis, J. Chromatogr., 559: 237-46 (1991). M. Ronaghi, M. Uhlén and P. Nyrén, “A sequencing method based on real-time pyrophosphate,” Science, 281: 363-365 (1998). D. A. Wheeler et al., “The complete genome of an individual by massively parallel DNA sequencing,” Nature, 452: 872-876 (2008). E. R. Mardis, “Next-generation DNA sequencing methods,” Annu. RevGenomics Hum. Genet., 9: 387-402 (2008). J. Shendure, et al. “Accurate multiplex polony sequencing of an evolved bacterial genome,” Science, 309: 1728-32 (2005). 12. J. M. Rothberg, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature, 475: 348-52 (2011). T. D. Harris, et al.,” Single-molecule DNA sequencing of a viral genome,” Science, 320: 106-9 (2008). J. Eid et al., “Real-time DNA sequencing from single polymerase molecules,” Science, 323: 133-8 (2009). S. Howorka, S. Cheley and H. Bayley, “Sequence-specific detection of individual DNA strands using engineered nanopores,” Nat. Biotechnol., 19: 636-9 (2001). F. J. Dyson, “Imagined Worlds,” Harvard University Press, Cambridge MA, 1998