A chemistry Professor at Indiana University, Bloomington, David Clemmer is a well-known figure in the world of analytical science. His work developing methods for the study of structures of complex low-symmetry in the gas phase has seen him recognized with several awards: the Fresenius Chemistry award and the John B. Fenn Distinguishing Contribution Award in 2018, to name just two.

Erin Baker completed her PhD in Chemistry at the University of California, Santa Barbara in 2005, before specializing in multi-dimensional separation techniques including LC, IMS and MS. Erin’s work focuses on evaluating molecular changes in biological and environmental systems.

A Scientific Fellow at Waters in Wilmslow, UK, Kevin Giles has been with the company for over 20 years. He has a PhD in Interstellar Chemistry. His work at Waters has primarily focused on the development of IMS-MS instrumentation, for which he was recognized in 2014 with the Industrial Analytical Science Award from the Royal Society of Chemistry.

What’s so special about ion mobility?
Erin Baker: IMS provides rapid structural separations based on the balance of two forces that impact the movement of an ion: the pull from the electric field and the drag force from collisions with buffer gas molecules. Variations on the electric field and stationary state of the buffer gas have given rise to multiple IMS techniques, including drift tube IMS (DTIMS), traveling wave IMS (TWIMS), trapped IMS (TIMS), field asymmetric IMS (FAIMS) and differential mobility analyzers (DMA). The different properties of each technique allow IMS devices to be constructed from low to high pressure values and have sizes ranging from just a few inches to many meters. Furthermore, each of these techniques can be interfaced with mass spectrometers to permit simultaneous acquisition of IMS structural and MS mass information on a rapid millisecond to second timescale. These multidimensional measurements are proving essential in assessing isomers and structural changes that cannot be evaluated with MS analyses alone.
David Clemmer: The timescale – millisecond measurements are slow enough that many types of experiment can be carried out, and fast enough that the observer doesn’t need to schedule more instrument time to make the measurements. Essentially, the mobility measurement is obtained for free (from a laboratory time perspective).
Kevin Giles: I would only add that the speed at which separation takes place (the peak capacity per unit time) allows effective sampling of temporally variant upstream processes such as chromatography or imaging.
IMS-MS was first developed in the 1960s – how has it evolved since then?
EB: IMS measurements have evolved throughout the last 60 years and become an increasingly important component of analytical analyses. One important development was the introduction of ion funnels to couple low-pressure IMS systems with MS in the late 1990s. This greatly increased the sensitivity of IMS-MS platforms by avoiding the losses associated with transferring ions between different regions, and thus enabled the rapid analysis of low-concentration compounds. Commercialization of these more sensitive IMS-MS instruments in the early 2000s gave a lot more groups access to the technology. Many groups found that the additional IMS dimension uncovered molecules missing or inseparable with MS-only analyses. These results have shown the value of the IMS separation and driven the commercialization of even more IMS-MS platforms.
DC: I entered the field in the 1990s, when I joined Martin Jarrold’s group at Northwestern University. At that time the structure of C60 was still unknown and our group was in a race with Mike Bower’s group to understand the structures of small carbon clusters. Great theoretical work and increased computing speed made it possible to calculate mobilities, and Jarrold and I were the first to couple ESI to IMS with the aim of resolving protein conformations. It would be hard to overstate how bad our initial measurements were! We would begin recording the spectrum early in the morning and not stop until early the next morning… And yet we only counted a few hundred ions in the largest peaks. But, compared with the calculated theoretical cross sections for simulated structures, it became clear that we were onto something exciting.
I set up my own group at Indiana University, focusing on IMS but a lot people thought we were wasting our time, asking “Who cares about the structures of proteins in the gas phase?” We responded by improving the measurement. Steve Valentine, Cherokee Hoaglund, Anne Counterman and I coupled ion traps to drift tubes to accumulate ions and then added a time-of-flight (TOF) mass spectrometer to make the first nested-IMS-TOF measurements. Jim Reilly and Ray Sporleder knew how to make TOF MS measurements and they helped us greatly. Milos Novotny told us to forget the model systems and work on a real sample, and his group taught us how to make tryptic digests. Before long we had 2D-IMS-TOF measurements of complex systems, including mixtures.
Soon, we had it all coupled to an LC and were able to run proteomics experiments by LC-IMS-MS and LC-IMS-collision-induced dissociation (CID)-MS. None of this worked very well! But we could resolve isomers and charge states and that was enough to get Kevin Giles and Tim Riley from Waters interested in developing it. IMS-IMS and circular instruments came along a little later and made sure that IMS was no longer seen as the poor relation of MS. We know now that there is a lot to learn from the ions before sending them into the MS.
KG: The availability of a commercial GC-IMS-MS system in the 1970s (made by PCP, West Palm Beach, FL) had a role in bringing the technology to more laboratories, but the real resurgence in interest began in the 1990s as key groups (Michael Bowers, Martin Jarrold, Herbert Hill, David Clemmer) began to study more biologically relevant species. Some of the biggest breakthroughs came from the Clemmer group. As David describes above, they showed nested LC-IMS-MS acquisitions on complex mixtures and improved sensitivity through ion trapping in sub-ambient pressure IMS and sequential IMS experiments followed by MS. Central to this advance was the utilization of TOF MS rather than quadrupoles. Another major contributor to the expansion of IMS-MS came in 2006 with the commercial availability of a quadrupole/TWIMS/TOF MS instrument, the Synapt HDMS from Waters.
In recent years, Dick Smith’s group have developed their structures for lossless ion manipulation (SLIM) approach towards extreme IMS resolving power (>1800) over separation path lengths of many hundreds of meters. Since 2013, two other companies have produced mainstream commercial IMS/quadrupole/TOF MS systems; the DTIMS-based 6560 from Agilent and the TIMS-ToF from Bruker. These instruments, combined with the Waters Vion (TWIMS/quadrupole/TOF MS) and Synapt systems, make IMS-MS more readily available than ever. The most recent addition to the commercial landscape is the Select Series Cyclic IMS instrument from Waters, released in 2019.
Where are we today?
DC: We have only scratched the surface of this technique. Buried in the broad peaks we generate today is a new type of vibrational spectroscopy that promises to tell us not only how many heavy isotopes are present, but also where they are located in each molecule. Dick Smith’s SLIM measurements and Waters’ cyclic instruments are beginning to reveal this information. Large ions remain mostly untouched. For example, IMS is enabling Vicki Wysocki’s IMS-SID and SID-IMS work on protein complexes. Exploiting fast gas-phase ion-molecules for all kinds of mixture analysis is another promising avenue – Steve Valentine leads the way here. The marriage of IMS with traditional chromatography and MS is ideal for addressing the emerging problems in the post-genome era. Fast computing and information management approaches present many new ways to interpret data and gain predictive power.

EB: Over the last decade the use of IMS in analytical measurements has rapidly increased, with applications in national security-related analyses, patient screening, environmental monitoring and numerous other areas. DTIMS, TWIMS, TIMS, DMA and FAIMS have all proved excellent tools for separating molecular classes and interfering ions, especially when coupled with MS analyses for the assessment of complex samples.
KG: IMS-MS has proven its worth in solving a wide range of analytical problems. Firstly, for analysis of complex samples, where the added peak capacity provided by IMS allows detection of lower-abundance species. Secondly, in screening applications, where CCS libraries (as well as the added peak capacity) can aid confirmation of the identity of target species – a particular area of focus has been in pesticide screening of foodstuffs. Thirdly, where measurement of CCS values in conjunction with calculated values allows structural elucidation. Compounds from small molecules to large protein assemblies have been studied, providing insight into areas such as isomeric heterogeneity of samples and structural biology.
In what applications will IMS-MS have the biggest impact in the future?
EB: While IMS-MS was once characterized as an emerging technique in the omics field, it is now well-integrated into modern omic measurements. Its utility in separating peptides and proteins with only minor structural differences has proven extremely powerful in bottom-up, middle-down, and top-down studies. IMS-MS has enabled the evaluation of isomers, which were previously inseparable or required long chromatography assays. Plus, the ability of IMS-MS to distinguish contaminant ions and molecular classes is proving essential across all omic analyses of complex environmental and biological samples. By measuring CCS values for available standards and compiling databases, rapid targeted analyses and identification of small molecules, lipids, glycans and peptides is possible. These CCS values are also being incorporated into computational methods, such as theoretical modeling of structures and machine-based approaches, enabling molecular insight when standards are not available.
KG: IMS is particularly powerful in conjunction with ambient ionization approaches, where the simplicity of sample introduction is augmented by the additional peak capacity provided prior to MS analysis. This is of particular benefit when real-time decisions need to be made, for example in food safety testing or histopathology. Another high-impact application is in screening, where the combination of peak capacity and CCS libraries can be used to increase sample throughput whilst reducing false positives and negatives, saving time and money.
By Robin H.J. Kemperman, Department of Chemistry, University of Florida, Gainesville, USA.
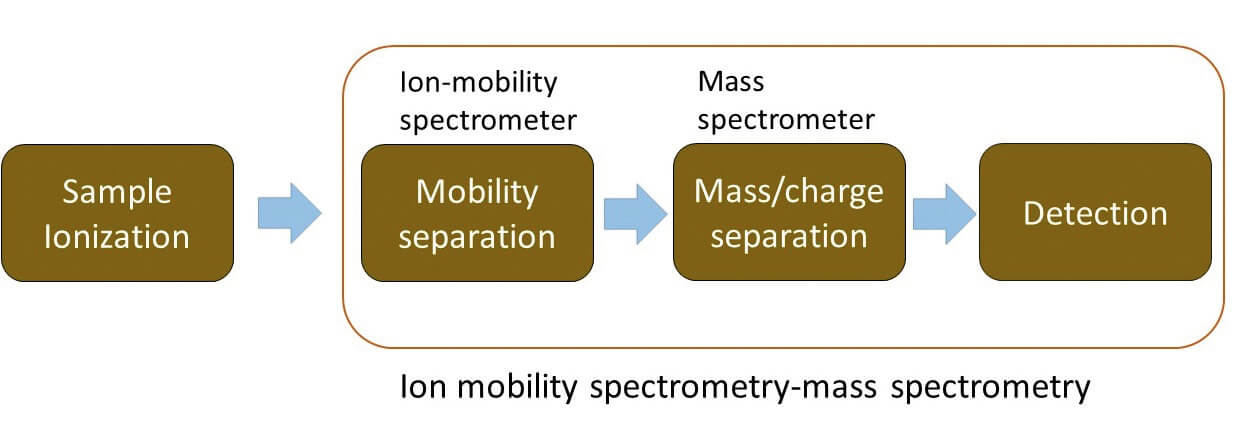
Ion mobility spectrometry (IMS) is an analytical technique that separates ions based on differences in their mobility whilst under the influence of an electric field and their interactions with a buffer gas.
While the use of IMS for analytical measurements and structural characterization is not novel, publications and conference proceedings over the past decade highlight a tremendous increase in IMS applications (1) (2). IMS has been successfully applied in a number of areas, including isomer separation, reducing analysis time in LC-MS, kinetic studies, structure elucidation, molecular modeling, and the compilation of collision cross section (CCS) databases.
Acronym overload?
There are a variety of different IMS platforms available commercially, with some important differences (and a wealth of acronyms).
Drift tube IMS (DTIMS) is the most traditional, in which ions are propelled through a series of stacked ring electrodes by an applied uniform electric field, undergoing interactions with a neutral buffer gas. Separation of the analytes is based on the size, shape, and charge of the ions. Larger or more extended ions have longer travel times compared with smaller and more compact gas phase ion structures due to larger drag forces. Using DTIMS, CCS values can be directly calculated from measured drift times, which is useful for compound identification and structure elucidation (3) (4).

Traveling wave IMS (TWIMS) moves ions forward in a buffer gas-filled cell in the direction of the detector. In contrast to DTIMS, a traveling or pulsed direct current (DC) voltage is applied, generating voltage waves that drive ions forward. In addition, a radio frequency (RF) voltage is used to confine the ions radially for higher transmission (5).
Trapped ion IMS (TIMS) separates ions based on a non-uniform electric field that increases towards the detector. Ions are pushed forward by a gas flow and, depending on the size-to-charge ratio, ions are trapped at different axial positions in the TIMS funnel while being radially confined by a RF voltage. When the electric field is decreased, ions are released towards the detector from high to low size-to-charge ratio (6).
Field asymmetric IMS (FAIMS) separates ions at much higher electric fields based on differences in mobility between high and low electrical fields. Ions with different mobilities have different trajectories and might strike one of the electrodes. Therefore, a DC compensation voltage is applied to shift the ion trajectories closer to the opposite electrode to detect specific ions of interest (7). FAIMS is a continuous IMS system without ion gates and is therefore well suited for use as an ion filter.
Keeping it clean
Despite the rich information that IMS-MS provides, most complex samples, such as biological mixtures, need some form of front-end separation prior to IMS-MS to remove interference.
LC, GC, and online extraction devices have been reported to be helpful in sample clean-up and reduction of ion suppression (8). Direct sampling techniques, such as matrix assisted laser desorption/ionization (MALDI), desorption electrospray ionization (DESI), liquid micro junction-surface sampling probe (LMJ-SSP), liquid extraction surface analysis (LESA), and laser ablation electrospray ionization (LAESI) have also been shown to benefit from a fast millisecond-based IMS separation prior to mass detection (9).
A valuable tool
IMS, in combination with direct sampling or font-end separation techniques, provides fast analysis whilst increasing the signal-to-noise ratio through background reduction. In addition, IMS provides isomer and isobar separation and can assign CCS values in some cases. These CCS values are reproducible for the same gas-phase structure on any IMS platform, in comparison to LC retention times, which can shift over time or change drastically when comparing different systems.
A loss in transmission occurs when an extra stage of separation is added to a workflow. This problem has been addressed in IMS-MS through the use of better ion guides and ion funnels, and improving the IMS duty cycle using multiplexing with DTIMS (8). For FAIMS, which often suffers from significant transmission losses, different geometries have been investigated (planar, cylindrical, hemispherical) to balance separation power and transmission (10). The resolving power of IMS has also been a limitation in some applications. To overcome this, multiple IMS cells have been coupled back to back, such as a FAIMS-DTIMS and TIMS-TIMS, to increase the peak capacity and take an IMS zoom of a previously isolated IMS peak. Structures for lossless ion manipulation (SLIM), which uses extremely long TWIMS path lengths on print plates, and cyclic TWIMS have been developed to increase the IMS resolving power dramatically (11) (12).
DC: I believe the most important future work will relate back to some of the early criticisms of gas-phase protein work. Because we all emerged from physics backgrounds, when we made the early measurements, we gave our critics more respect than they probably deserved. Gas-phase proteins are often solution structures that become trapped upon removal of solvent in configurations that do not reflect native structures. In the early days, everyone was focused on visualizing native structures, so this was seen as undesirable. But what we are seeing is dozens of new non-native structures for proteins that cannot be observed with any other approach. A “cooperative, two-state” transition is really a composite of many structures. IMS-MS measurements appear to provide more information about the non-native states than any other method. If we are really going to understand protein structure – perhaps the most important problem in biology – we will need to understand the role of non-native structures. How stable are they? What are their lifetimes? Do they aggregate?
What are your predictions for the future?
DC: MS methods didn’t come about overnight. There was nearly a century of work before we saw Alan Marshall’s high-resolution measurements. After that came Alex Makarov’s Orbitrap and suddenly everyone could make MS measurements easily and without cryogens. High-resolution IMS-MS has only been around for decade or so. In 50 years, it will have found its place and we won’t think so much about the difficulties. The new cyclic instruments allow us to carry out IMSn experiment – enabling an exciting new approach for state-to-state-(to-state)n experiments.
KG: There have been significant advances in transmission and resolving power in the past two decades. The challenge is that higher resolution requires longer separation times and to maintain performance (both transmission and resolution), the ion optics need to have increased charge capacity, mandating larger devices. Multiplexing approaches have proven useful in this area but are not without limitations. Another key challenge is to further improve the accuracy of CCS determination, both through instrumental measurement and theoretical calculation. These two requirements go hand-in-hand if more subtle structural differences are to be unraveled between species and if cross-platform CCS databases are to be produced. Instrument precision and reproducibility is, however, generally good, facilitating platform-dependent screening applications. One of the biggest issues has been a lack of software support for mobility data analysis. Significant inroads have been made in this area but there is still plenty of scope to ensure IMS-MS is accessible and embed it into routine workflows.
EB: Undoubtedly, the utility of IMS-MS hinges on increasing its resolving power and successfully coupling it with other pre-separation, fragmentation and software tools. Fortunately, developments in all these areas are taking place to enhance the capabilities of IMS-MS measurements. Higher-resolution IMS and MS instrument platforms, such as the SLIM approach Kevin and David mentioned earlier, are becoming available and provide greater confidence in global measurements of complex biological samples. New fragmentation approaches such as electron-capture dissociation are being added to various instrumental IMS platforms, and software tools enabling rapid analyses are also being developed to push the technology further. The analysis of the multi-dimensional LC-IMS-MS data has proven difficult, due to the many measurement dimensions (LC, IMS, MS and data-independent or data-dependent acquisition MS/MS). Despite this, the future seems promising. The incorporation of numerous software packages including the open source Skyline software means that the capabilities of IMS-MS are only expected to increase in the future, making it an essential tool for high-quality sample analyses.
KG: I think the next few years are going to be extremely interesting for IMS, particularly in application development. There have been huge strides made in hardware capability and the application of this added horsepower to more challenging or complex studies is providing more detail than previously possible. Simply increasing IMS resolution will not provide all the answers. The added flexibility and specificity of IMSn, in the form of either sequential IMS devices or multipass cyclic IMS instruments, will be particularly enabling. I feel the broader acceptance of IMS-MS will come from its increasing use as a routine tool to provide better/quicker answers to analyst’s questions. This will require ongoing development of software and integrated hardware.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- JC May, JA McLean, “Ion Mobility-mass spectrometry: time-dispersive instrumentation”, Anal Chem, 87, 1422-1436 (2015). DOI: 10.1021/ac504720m.
- S Armenta et al., “A review of recent, unconventional applications of ion mobility spectrometry (IMS)”, Anal Chim Acta, 703, 114-123 (2011). DOI: 10.1016/j.aca.2011.07.021.
- SM Stow et al., “An interlaboratory evaluation of drift tube ion mobility mass spectrometry collision cross section measurements”, Anal Chem, 89, 9048-9055 (2017). DOI: 10.1021/acs.analchem.7b01729
- JC May et al., “Ion mobility collision cross section compendium”, Anal Chem, 89, 1032-1044 (2017). DOI: 10.1021/acs.analchem.6b04905.
- BT Ruotolo et al., “Ion mobility-mass spectrometry analysis of large protein complexes”, Nat Protoc, 3, 1139-1152 (2008). DOI: 10.1038/nprot.2008.78
- ME Ridgeway et al., “Trapped ion mobility spectrometry: a short review”, Int J Mas Spectrom, 22-35 (2018). DOI: 10.1016/j.ijms.2018.01.006
- AB Kanu et al., “Ion mobility-mass spectrometry”, J Mass Spectrom, 43, 1-22 (2008). DOI: 10.1002/jms.1383
- X Zheng et al., “Coupling front-end separations, ion mobility spectrometry, and mass spectrometry for enhanced multidimensional biological and environmental analysis”, Annu Rev Anal Chem, 10, 71-92 (2017). DOI: 10.1146/annurev-anchem-061516-045212.
- M Sans et al., “Advances in mass spectrometry imaging coupled to ion mobility spectrometry for enhanced imaging of biological tissues”, Curr Opin In Chem Biology, 42, 138-146 (2018). DOI: 10.1016/j.cbpa.2017.12.005.
- R Cumeras et al., “Review on ion mobility spectrometry. Part 1: Current instrumentation”, Analyst, 140, 1376-1390 (2015). DOI: 10.1039/ c4an01100g.
- L Deng et al., “Ion mobility separations of isomers based upon long path length structures for lossless ion manipulation combined with mass spectrometry”, Chemistry Select, 1, 2396-2399 (2016). DOI: 10.1002/slct.201600460.
- C Eldrid et al., “Gas phase stability of protein ions in a cyclic ion mobility spectrometry traveling wave device”, Anal Chem, 91, 7554-7561 (2019). DOI: 10.1021/acs.analchem.8b05641.



