The entry of next-generation sequencing (NGS) into clinical practice has been disruptive. We can now analyze many more samples, with much less money, and in more depth than ever before, allowing us to comprehensively interrogate the entire genome of cancer cells. In the past, we could only assess one known gene at time, but now NGS allows us to look at the entire genome in a single assay – completely changing how we do translational research and how we will do clinical genomic testing. Traditionally, we have taken a bottom-up approach in biomarker discovery. Basic scientists look at an interesting pathway in the cell that may be associated with tumor behavior. They find a limited number of genes or proteins related to that pathway, and study them to find out how they work, and whether they might have potential as biomarkers. Along with John Cheville, I direct the Biomarker Discovery Program at the Mayo Clinic’s Center for Individualized Medicine. Here, rather than starting from a gene or protein of interest, we start with a practical clinical question that fills a physician and patient need, and aim to identify a biomarker and develop a test to answer that clinical need. We refer to this process as product-driven biomarker discovery. Economically, we believe this makes a lot of sense – the clinical need dictates the experimental design, validation and assay development, rather than a more random approach.
The search is on
At the DNA level, cancer acquires mutations and small indels (insertions or deletions of bases) or large chromosomal rearrangements. A lot of investment has gone into investigating point mutations but, around seven years ago, I decided to turn our attention towards large chromosomal alterations, which occur in many cancers and are important in determining cancer behavior and response to treatment. But how can you best identify those alterations? One approach is whole-genome analysis, but at the moment this would cost more than $10,000 per patient. And before you can find any commonalities among patients, you have to do hundreds of samples, so that whole-genome analysis quickly becomes too expensive. It also generates a huge quantity of information that we don’t always know how to handle, and produces a lot of “noise”. And as the data grow and grow, we need faster and better algorithms, which can be a challenge to create.To solve this problem, we developed a new strategy based on Illumina’s mate pair sequencing. Mate pair sequencing allows us to look at the whole genome of tumor cells for rearrangements, deletions, amplifications, and gains – all for less than $1000 per sample. Our protocols and algorithms make the technique viable for routine clinical use. Typically, if you contaminate a tumor sample with too many normal cells, you lose the signal from the cancer cells. So we have developed protocols for laser-capture microdissection to obtain a very pure population of cells. In addition, we developed bioinformatics algorithms we call BIMA and SVA tools that filter out the “noise” and significantly reduce false-positive results. In brief, BIMA handles sequencing artifacts inherent in mate pair library preparation, including biotin junction reads, paired-end read contamination, chimeras, and so on. With these new protocols and algorithms, mate pair sequencing is being implemented in our clinical laboratories, where we believe it will replace 95 percent of conventional cytogenetic testing. For instance, the complex multiple tests applied to bone marrow samples in patients with leukemia can now be done with a single mate pair sequencing assay – faster, cheaper and comprehensive (1)(2).
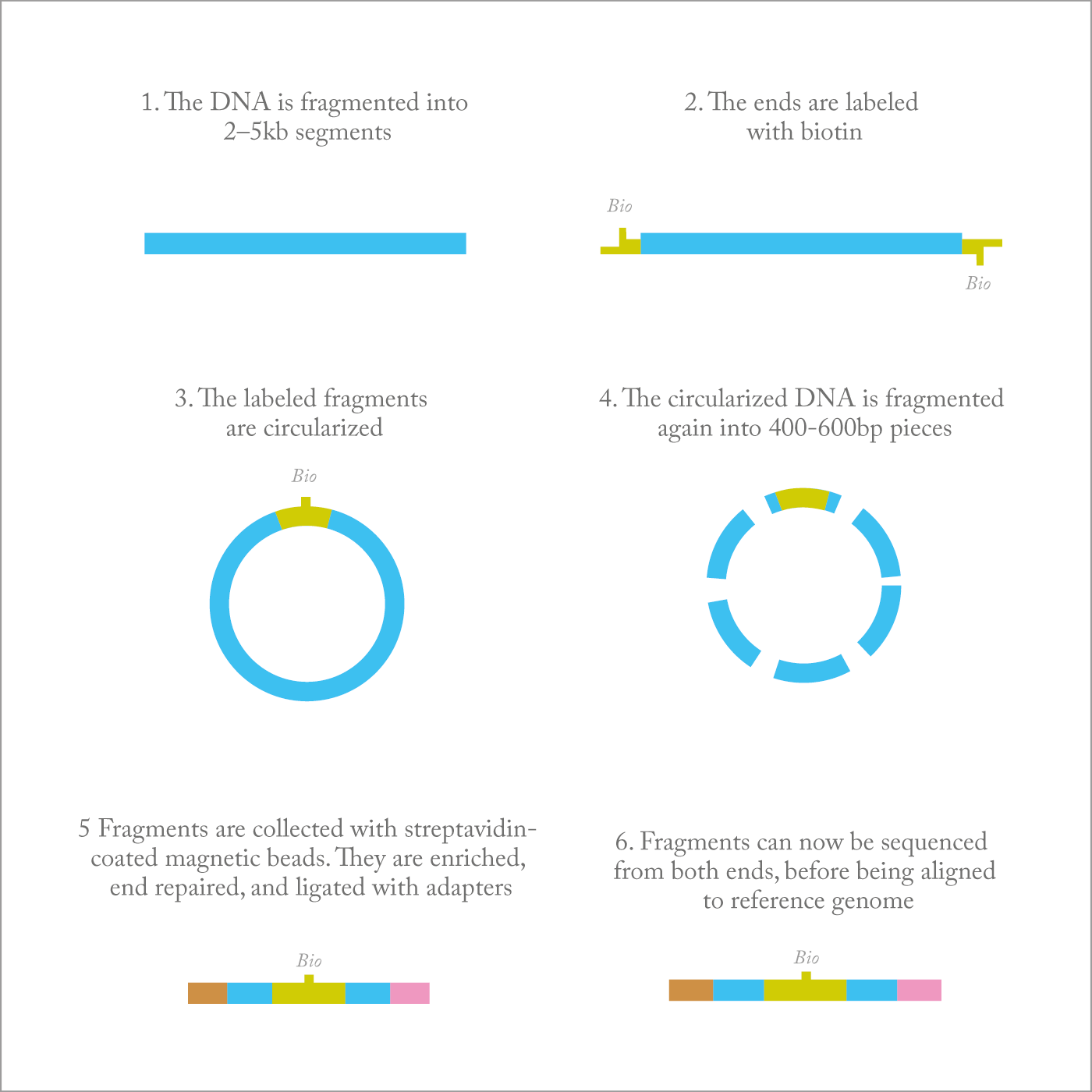
The tradeoff for the broad coverage and low cost of mate pair sequencing is that it has limited sensitivity, so there will still be a role for more sensitive assays such as fluorescence in situ hybridization (FISH). It’s a little like Google Earth – mate pair sequencing allows you a bird’s eye view of the entire genome but if you want to see all the details you have to focus and zoom. We envision that mate pair sequencing will be the “go to” test, with karyotyping, FISH, and arrays being used as needed for confirmation or follow-up.
Sequence, and ye shall find
The first clinical oncology application is likely to be in hematological cancers, in which rearrangements and gene fusions are already being used as biomarkers to aid diagnosis and direct treatment. In this case, mate pair sequencing can simply be incorporated into existing protocols to provide a more cost-effective means of detection. More challenging, but very exciting, is our ongoing work on solid tumors. There are several important clinical questions that we believe mate pair sequencing can help resolve. For example, the majority of men will develop some form of prostate cancer as they age. In most cases the disease will be slow growing and require no treatment, but some will be much more dangerous, fast-growing tumors, with a risk of metastasis. Currently, it is difficult to tell the two types apart, leading to unnecessary treatment for many men. We are using mate pair sequencing to search for genomic markers that can separate clinically insignificant prostate cancer from more aggressive prostate cancer that requires treatment (3)(4). Lung cancer is another area of interest for us (5). Some patients present with more than one tumor in their lungs; it is important for physicians to know whether this is the result of one tumor that has metastasized or two separate primary tumors. Gene rearrangements are common in lung cancer (for example, the fusion of EML4 and ALK to form the EML4-ALK oncogene) and tumors of common origin will have identical rearrangements and breakpoints. Using mate pair sequencing, we are able to detect similarities and differences in the rearrangements of the two tumors, and determine if they are related. These results will determine if the patient is a candidate for curative surgery for two independent primary tumors or should receive chemotherapy for metastatic lung cancer.Making it easy
The people who will be using these tests – clinicians, researchers and pathologists – understand the disease and its genetics very well. What they don’t necessarily understand is data. And the data that results from mate pair sequencing is completely different to the FISH panels or chromosomal bands that they are used to. A bigger challenge for users is that the technologies they used in the past gave them access to a relatively small amount of information. Today, whole-genome technologies, such as mate pair sequencing, are likely to result in information overload. And it’s not just too much information, it’s too much important information. The next big challenge for us is to create visualization techniques to transform data into something that can easily be understood by clinicians and patients. Genomic research programs are expensive enterprises, so we need a return on investment if we are to continue our work. But, for me, financial reward has never been a motivation – our primary goal is to improve patient care. And it’s clear that genomics – using the right tools – will play an important part in achieving that. It is a very satisfying feeling to know that we have helped drive mate pair sequencing into the clinic to improve patient care. George Vasmatzis is co-director of the Biomarker Discovery Program within the Center for Individualized Medicine at the Mayo Clinic, Rochester, MN, USA.Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- G Vasmatzis et al., “Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas”, Blood 120, 2280–2289 (2012). AL Feldman et al., “Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing”, Blood 117, 915–919 (2011). SJ Murphy et al., “Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer”, DNA Research 19, 395-406 (2012). IV Kovtun et al., “Lineage relationship of Gleason patterns in Gleason score 7 prostate cancer”, Cancer Res 73, 3275–3284 (2013). SJ Murphy, “Identification of independent primary tumors and intrapulmonary metastases using DNA rearrangements in non-small-cell lung cancer”, J Clin Oncol 32, 4050–4058 (2014).



