Diverse domains of analysis, including food safety, environmental, clinical, forensic toxicology and doping control, face a similar, and substantial, challenge.
Take food analysis, for example. A foodstuff may contain one, two or multiple residues of pesticides, veterinary drugs, natural toxins (mycotoxins, plant toxins) environmental contaminants, packaging contaminants and processing contaminants. Within each of these categories, hundreds of substances are known. For many of these chemicals, legal limits in food commodities have been established; for others, limits have not been set, either because authorities are unaware that they occur in food or because the toxicant is not yet recognized as such.
In environmental analysis, the number of chemicals detected in surface water is increasing. These range from well-known persistent pollutants and pesticides to pharmaceuticals and metabolites that elude the sewage treatment system. Many of them pose a risk to aquatic life and to our drinking water resources. Attempts are being made to generate a list of key chemicals to be monitored but this is hampered by a lack of data on their occurrence. (Of course, in an ideal world, everything should be assessed.) Analysts from toxicology and doping control laboratories face a similar situation when analysing urine, oral fluids or blood samples for toxic substances and illicit and designer drugs. What is shared across all of these domains is a requirement to detect and determine very high numbers of substances, known and unknown, in a wide variety of sample matrices, at levels ranging from sub µg/kg to low mg/kg level. It’s a huge analytical challenge. At present, organic trace analyses are conducted as targeted methods (see “Past and Present - Targeted Measurement” at the end of this article). But the future looks rather different.
Using the classical, targeted approach, you must decide what you want to know, set up the method and perform your analysis. In contrast, a non-targeted approach analyses everything, then you decide what you want to know and extract the information from the raw data. Table 1 summarizes the targeted and non-targeted approaches. Non-targeted measurement uses generic sample preparation and chromatography, combined with full-scan mass spectrometric detection. All ions are detected during the entire chromatographic run time and, in contrast to MS/MS, there are (in principle) no limitations to the number of substances that can be detected (see Figure 1). Using non-targeted measurement, you may search the raw data of previously measured samples for analytes that were not of interest or not known at the time of analysis, without re-sampling or re-analysis of stored samples. Another feature is that a fingerprint, known as a total ion chromatogram (TIC), is obtained for each sample. This can be used for comparison with existing sample profiles to reveal deviations and/or to aid in the identification of unknowns. The potential of non-targeted measurement was recognized long ago; the approach as such is not new. Applications were reported in the 1980s for the toxicological screening of drugs, and soon afterwards for detection of pesticides residues in food and of priority environmental pollutants in surface water. These early applications were based on GC-EI-MS (single quadrupole full scan) or GC-EI-ITD (ion trap detection). However, many substances not amenable to GC were not covered, and for those that were, the sensitivity and selectivity were limited. To improve selectivity, two approaches have been pursued. One is to increase chromatographic resolving power using comprehensive two- dimensional GC (GCxGC). This reduces peak overlap, resulting in cleaner mass spectra or less interference of the diagnostic ions. Since GCxGC refocuses the chromatographic peak, sensitivity is also improved although, since these peaks are very narrow (~0.1 s), fast-scanning (>100 Hz) time-of-flight (TOF) mass analysers are required. The second approach is to increase the resolving power of the mass spectrometer. Selectivity is improved because ions that have the same nominal mass but a different exact mass can be separated, again resulting in reduced interference of diagnostic ions. However, the GC-EI-hrTOF-MS instruments of the mid 2000s offered only intermediate resolution (5,000-7,000 FWHM) which meant that they suffered from limited dynamic range and unreliable mass accuracy. Only very recently have new instruments with better performance characteristics become available (13,000-25,000 FWHM).
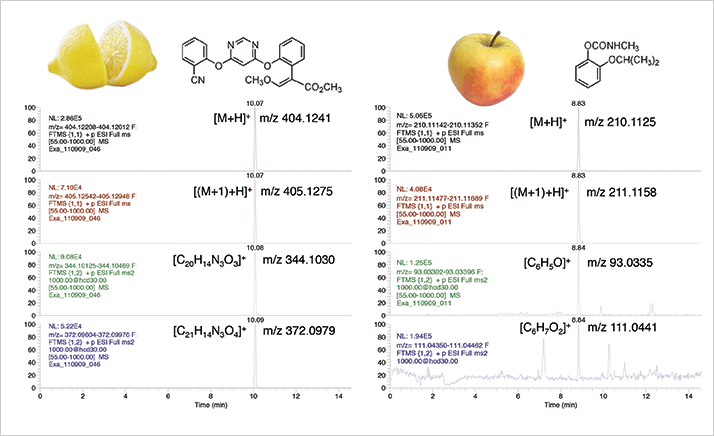
Initially, high resolution TOF systems for LC presented these same shortcomings. However, dynamic range, sensitivity and selectivity (resolving power/mass accuracy) have all improved dramatically over the past eight years. Resolving powers of 25,000-50,000 are the norm, while the benchtop Orbitrap MS systems go up to 140,000. Direct comparisons between high resolution MS and triple quadrupole MS/MS are not easily made. Roughly speaking, they are comparable in terms of selectivity when the resolving power is ~50,000 or higher (2) and, while high resolution MS falls short of the newest triple quadrupoles in terms of sensitivity, it is fit-for-purpose for most applications. Fragment ions to aid identification can be generated in the source or in a collision cell using alternating scan events and, when done without precursor ion selection, there are no compromises with respect to non-targeted measurement (see Figure 2). Given the quantitative performance of current instruments, even for targeted applications, full-scan measurement will be the method of choice in the near future because the measurement is more straightforward compared with MS/MS.
Developments in hrMS detectors for LC have resulted in them being coupled to GC using alternative ion sources (APCI, atmospheric pressure chemical ionization). This soft ionization technique is somewhat less generic than EI, but the abundance of molecular or protonated ions combined with sensitivity and resolving power make it an attractive possibility (4). The potential of non-targeted measurement by chromatography with full scan MS has been demonstrated in numerous papers in food safety (2), environmental analysis (5) and clinical, forensic toxicology and doping control (6,7).
Challenge 1: automated analyte detection Untargeted measurements provide an overwhelming amount of data. For each substance, including analytes of interest but also all matrix compounds, information is obtained on retention time(s), accurate mass of ions (m/z 50-1000), isotope patterns and/or spectra, and intensity (from noise to saturation). The question is, how do you find what you’re looking for in all this raw data? In most applications, the analyst has a long target list of substances in mind, and interrogates elemental composition (provided by the exact mass and isotope signature) or an EI-spectrum. Although the measurement is untargeted, this approach, in the end, is equivalent to a targeted method but for a very high number of targets. A consequence of this is that analyte detection has to be automated; manual evaluation of spectra or extracted ion chromatograms would be too time-consuming in routine practice. In the case of GC-EI-MS, spectra are more-or-less instrument-independent and large libraries are available. The instrument software performs searches of sample spectra against library spectra, although few packages are specifically designed for automated detection and reporting on larger numbers of samples and various in-house solutions have been described in the literature. With high-resolution MS data, the detection of analytes is based on a signal for the exact mass of one or more diagnostic ions at the expected retention time. Here, at least part of the relevant information for analyte detection is dependent on the instrument and experimental conditions, which complicates the establishment of comprehensive reference databases. At the moment, databases are still a work in progress, for both vendors and users. The initial detection of a spectrum, signal or peak is performed by a software algorithm either directly on the raw data or after a pre-processing step. Often, a number of parameters can be set to influence peak detection. Once a peak is detected, tolerances are set on spectral matches, isotope fits, mass deviations, retention time, intensity thresholds, and so on, to ensure that the number of false positives and false negatives is acceptable. In practice, optimization of software parameters and thresholds is the most time-consuming part of the method development, especially when sensitivity is critical. New, instrument-specific software packages and updates are regularly released by vendors, which requires re-optimization of parameters. This has triggered some researchers to develop their own software solutions (8). Whereas the hardware is fit-for-purpose, the lack of comprehensive databases and inadequate software for automated detection are major bottlenecks hindering the widespread application of untargeted measurement for wide-scope screening at the moment. Challenge 2: validation Another challenge faced by non-targeted measurement aimed at the detection of very high numbers of analytes is method validation. To illustrate, consider veterinary drugs in food of animal origin and pesticides in food and feed. EU-guidance documents were established in 2010 (3), under which a method is considered fit-for-purpose when a substance can be detected in 95% of the samples (with N≥20 in an initial validation exercise that needs to be complimented by on-going analytical quality control). Because even similar substances can behave very differently when analysed by chromatography-MS, the guidelines require validation to be performed at the individual analyte level. We have carried out two validation studies for a total of 350 pesticides measured by GCxGC-TOF-MS and HPLC-Orbitrap-MS. The criterion was met for the majority of the tested pesticide/matrix combinations at levels down to 0.01-0.05 mg/kg. The constraint here is not so much sample analysis and validation as having all of the standards available and maintaining the collection: 350 pesticides is not comprehensive and represents only a small part of the potential of the method. These new analytical possibilities throw up new legislative and ethical issues. For example, if a sample of wheat taken to verify compliance with maximum limits for mycotoxins detects an illegal pesticide, can enforcement actions be taken for the latter? When urine samples are taken for biomarker analysis to investigate exposure to mycotoxins, but at the same time metabolites of drugs-of-abuse are found, what should be done with this information?
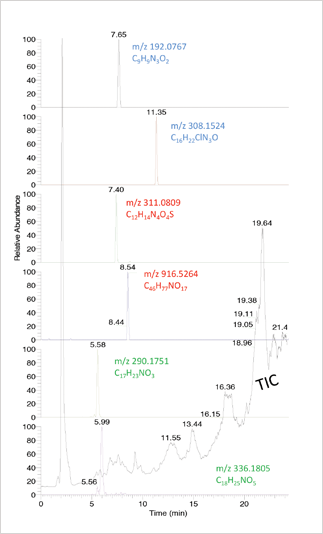
Currently, one way of identifying unknown substances is by analysing features; for example, the sample under investigation may share a fragment typical for a certain class of substances, or a characteristic chlorination or bromination isotope pattern (see Figure 3). High resolution/accurate mass data are ideally suited for this work, but with proper scripting software it can also be done using full-scan GC-EI-MS data.

The next step in data evaluation is non-targeted detection. Using GC-EI-MS, the spectrum for each peak can be run against a generic library of more than 200,000 spectra. Since the number of resolved peaks in a sample may be in the thousands and one spectrum may match reasonably well with multiple reference spectra, there may be various candidates that require further investigation. Using soft ionization and high resolution MS, an elemental composition derived from the accurate mass can be run against compound databases such as ChemSpider or PubChem Compound. Here, the number of candidates may be higher still, since one elemental composition may correspond to a lot of chemical structures. Fragmentation data are then needed to aid identification. A special case of non-targeted detection is provided by comparative studies in which fingerprints from non-deviating (non-treated) products are compared with fingerprints from deviating products. Multivariate analysis of the fingerprints may reveal certain peaks that are different in the two categories and which may then be focused upon for identification. A good example of this is the metabolomics approach to detect cattle treated with synthetic hormones (9). Similar types of approaches are being explored to assess the chemical safety of food (10).
Non-targeted measurement using full-scan high resolution MS is rapidly becoming fit-for-purpose for the determination of small molecules at low levels in a wide variety of matrices. It allows us to do the things that would otherwise require triple quadrupole MS, namely quantitative targeted detection, in a more straightforward way. At the same time, it offers many more new possibilities for targeted and untargeted screening of other analytes of interest, which can be done directly after the analysis or at any later point in time. Improvements in software to digest the enormous amount of data generated have been made in the past few years, but more efforts are needed to take full advantage of the potential. I foresee that for many applications, non-targeted measurement (full scan MS) will gradually replace targeted measurements (triple quads). Hans Mol is Team Leader, Contaminants & Toxins at RIKILT, Wageningen UR, The Netherlands.
In targeted measurement, the goal is set before analysis: “we wish to determine substance X in matrix Y”. Next, a method is selected or developed to provide a quantitative measurement. The procedure has three steps: (1) sample preparation, which consists of extraction, clean up and optional derivatization; (2) chromatographic separation, and (3) detection. All three contribute to selectivity and sensitivity. In the early days (1970s–2000), the selectivity of detectors included element-selective detectors for GC, and UV and fluorescence detectors for LC. The limitations of these systems were compensated for by extensive clean-up procedures that selectively isolated certain analytes or analyte classes. This inherently-restricted analysis changed with the introduction of MS detectors. In GC, single quadrupole and ion trap MS enabled generic and reasonably selective detection: generic because electron ionisation (EI) works for virtually any substance that is GC-amenable, and selective because ions are obtained that are often highly diagnostic for the analyte of interest. The improved selectivity in instrumental analysis made it possible to reduce clean-up efforts during sample preparation, retaining more classes of analytes in the final extract. This in turn offered a substantial increase in the number of analytes that could be determined by one method. GC-MS was the dominant technique for multi-analyte determination for a decade (1990s). Then came the coupling of LC to triple quadrupole MS (MS/MS). This dramatically improved selectivity and sensitivity, allowing further simplification of sample preparation and eliminating the need for derivatization. Today, sample preparation is essentially non-targeted, being reduced to extraction and removal of the bulk matrix, and dilute-and-shoot approaches (1). LC-MS/MS predominates, complemented by GC-MS/MS where required. The overall measurement is still targeted, because with MS/MS acquisition, analyte-specific transitions are measured. Furthermore, there are limitations in the number of transitions, that is, analytes, that can be targeted simultaneously in one method (currently several hundreds). Consequently, a range of (very similar) methods are used, often focusing on certain types of analytes.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.

References
- H.G.J. Mol et al., “Toward a generic extraction method for simultaneous determination of pesticides, mycotoxins, plant toxins, and veterinary drugs in feed and food matrices”, Anal. Chem., 80, 9450-9459 (2008). A. Kauffmann, “The current role of high-resolution mass spectrometry in food analysis”, Anal. Bioanal. Chem., 403,1233–1249 (2012). H.G.J. Mol et al., “ Guidelines for the validation of qualitativemulti-residue methods used to detect pesticides in food”, Drug Test Analysis, 4 (Suppl. 1), 10–16 (2012). T. Portoles et al., “Potential of atmospheric pressure chemical ionization source in GC-QTOF MS for pesticide residue analysis”, J. Mass. Spectrom., 45, 926–936 (2010). F. Hernández et al., “Current use of high-resolution mass spectrometry in the environmental sciences”, Anal. Bioanal. Chem., 403, 1251-64 (2012). A.H.B. Wu et al., “Role of liquid chromatography high-resolution mass spectrometry (LC-HR/MS) in clinical toxicology”, Clin. Toxicol., 50, 733-742 (2012). R.J.B. Peters, et al., “Screening in veterinary drug analysis and sports doping control based on full-scan, accurate-MS, Trends Anal. Chem., 29, 1250-1268 (2010). A. Lommen, “MetAlign: interface-driven, versatile metabolomics tool for hyphenated full-scan MS data preprocessing”, Anal. Chem., 81, 3079–3086 (2009). J.W.C. Rijk et al., “Metabolomics approach to anabolic steroid urine profiling of bovines treated with prohormones”, Anal. Chem., 81, 6879–6888 (2009). J.P. Antignac et al., “Mass spectrometry-based metabolomics applied to the chemical safety of food”, Trends Anal. Chem., 30, 292-301 (2011).



