Why does the analysis of proteins remain so important?
Andrea Gargano: The analysis of proteins is essential for understanding the complexity of the communication that takes place in our bodies. Improving tools for protein analysis has important implications for medical science, where we aim to understand the mechanisms of action of bioactive molecules, find (bio)markers for diseases, and characterize new classes of pharmaceuticals (for example, antibodies). Developments in protein analysis promote research at the boundary between biology and chemistry; namely, biochemistry, system biology and bioengineering. Moreover, recent progress in proteomic research has demonstrated that advanced analytical tools for protein analysis open up new possibilities in fields beyond protein science, such as polymer and biopharmaceutical research. In essence, protein analysis is important because it is an analytical challenge with big implications. Koen Sandra: Proteins have many functions – structural (keratin in hair, collagen in bones, skin), mechanical (myosin/actin in muscle movement), transport (hemoglobin for oxygen transport in blood), defense/immune (antibodies), biochemical reactions (enzymes), hormones (insulin regulated glucose metabolism) – and the list goes on. Amongst many other benefits, analysis of proteins can lead to the discovery of novel drug targets and biomarkers for disease diagnosis, prognosis, and prediction, and is key in the concept of personalized medicine. Proteins themselves are also on the rise as therapeutics; hence, from a biopharma perspective, accurate analysis is essential. Shabaz Mohammed: If it’s not already clear from Koen and Andrea’s answers, I’ll add that a significant number of diseases, including many types of cancer, can be related to the dysfunction of proteins and their interactions. Thus characterizing their structure, function and interactions is of the utmost importance. John Yates III: Proteins are the operational agents of cells. They form structures, they transmit signals, they catalyze reactions to form metabolites, they form protein complexes. If you want to know how cells work, you have to study proteins.How far are we from characterizing the proteome of complex organisms?
AG: We are a long way off – potentially 100,000 proteoforms (90 percent) away from the entire proteome of a complex living organism (including ourselves). However, important results have been achieved with current technology, supporting genomic and transcriptomic results and enabling discoveries in biomarker research. So far, we are just capable of characterizing the tip of the proteo-berg. To better grasp the proteome complexity, we need better analytical as well as chemometric and statistical tools that are capable of refining and rationalizing the large amount of data that we are collecting. SM: The completion of genomes and the massive improvements in the last decade in cell manipulation, protein chemistry, chromatography and mass spectrometry allow one to identify pretty much any protein in a cell. There are still a few exceptions, such as very low copy number proteins or those with ‘difficult’ physicochemical properties (for example, hydrophobicity in the form of transmembrane domains) – and there are some proteins that suffer from both sets of problems, such as olfactory receptors. Given enough time and resources, one can generate evidence of presence for pretty much all the proteins in a particular type of cell. However, it isn’t something that is often performed because the resources and time required to carry out such a feat are huge – and there aren’t really any good reasons to carry out such an experiment. We often limit ourselves to a few days’ mass spec time for an experiment and the depth level achieved (approximately 8-10,000 protein families for human cells) is sufficient for most biological questions. Consequently, the jump to characterizing all the distinct types of cells in an organism, of which humans have hundreds, is a far off dream. JY: I’d say we’re pretty close to being able to identify the presence of all proteins in a mammalian cell. But the difficulty is knowing how many proteins are really there. After all, protein expression varies with conditions. Additionally, the complete proteome will encompass protein isoforms and modified forms for all expressed genes – and that is a pretty large set of proteins.What are the strongest arguments for using 2D-LC in proteomics?
KS: Why has proteomics historically been performed using 2D-PAGE instead of 1D SDS-PAGE? Because one can identify and quantify many more proteins. The same conclusions can be drawn when evolving from 1D to 2D-LC in proteomics. The inability of 1D-LC to adequately resolve real-world mixtures of high complexity is the driving force behind using multidimensional separations. It is no surprise that the field of proteomics is widely adopting the technology given the enormous sample complexity encountered. Simple unicellular systems already contain thousands of proteins, and for now, one can only speculate on the complexity of the clinically valuable and most complex human serum/plasma proteome. Due to the preferred handling of peptides over proteins and the consequent proteolytic digestion of proteins prior to downstream processing, every protein is represented by dozens of peptides. It is not unheard of to be confronted with thousands of peptides, spanning a wide concentration range, that have to be introduced into the mass spectrometer in a way that allows successful qualitative – and quantitative – measurement. Multidimensional LC possesses the additional resolving power to substantially reduce the complexity of such peptide mixtures and, therefore, to increase the number of measurable peptides, to widen the overall dynamic range and consequently to increase proteome coverage. In addition, the implementation of an extra separation dimension has also been shown useful in the targeted analysis of proteins. In contrast to the above described discovery proteomics approach, targeted analysis does not require the analysis of the entire first dimension. AG: State-of-the-art 1D-LC chromatographic setups, using long columns (0.5 to 1 m), packed with materials of small particle size (2 µm) and shallow gradients (from 1 to 6 hrs) can provide peak capacities up to 1500. However, the components of proteomics samples are hundreds of thousands (if not millions) and thus, during LC-MS analysis, mass spectrometry instruments have to deal with complex mixtures of peptides present in vastly different concentrations. Co-elution leads to ion-suppression effects that, together with MS dynamic range limitations, compromise the analysis of low-abundant species and thus limit the depth of proteomic investigations. As Koen notes, online comprehensive two-dimensional liquid chromatography (LC×LC) enables deeper investigations of the proteome because of its higher resolving power. Moreover, LC×LC can combine chromatographic methods with different retention mechanisms (such as ion exchange, hydrophilic interaction LC, or high pH reversed-phase × low pH reversed-phase). As a result, sample components can be separated with orthogonal selectivities. In addition, the fast and efficient second dimension separations enabled by UHPLC technology significantly reduce the peak widths (typically ranging from below 1 to 10 s) enabling higher peak capacities with respect to 1D-LC (more than 1000 in less than 1h). SM: I can echo Koen and Andrea. The first step in most protein characterization experiments involves chopping up proteins into peptides. To comprehensively identify a human proteome, one needs to handle a peptide mixture numbering in the millions that span a dynamic range of 7-9 orders of magnitude. The pace of improvement in 1D LC-MS is astonishing; however, issues remain and there are certain hard limits that haven’t been addressed. The latest mass spectrometers have (successful) sequencing rates of around 10-20 peptides per second and, with state of the art UHPLC systems, one can hope to identify 20-40k peptides in a single analysis. Increasing those numbers won’t be easy since the dynamic range of a mass spectrometer is (at best) five orders and current MS systems can just about sample peptides at the bottom of its restricted dynamic range. Increasing speeds don’t really improve the situation since the mass spectrometers can’t collect sufficient populations of the low abundant peptides for a successful sequencing event because of the limited dynamic range. It’s also been predicted that the resolving power of current UHPLC systems improvements would plateau at around double current values. Thus, the only way to increase capacity is to fractionate and the most powerful and sensitive (by far) method of fractionation is an additional round of chromatography. JY: To get the complete mammalian cell proteome as described above will require a tremendous peak capacity and it is unclear if 1D LC can deliver that level of performance. Moreover, developments occurring in ultra-high resolution 1D LC, of course, can be adapted for 2D LC.Andrea Gargano is a post-doctorate researcher at the University of Amsterdam, specializing in two-dimensional liquid chromatography. In the summer of 2015, Gargano was awarded a Veni grant from the Netherlands Organization for Scientific Research (NWO) and he is currently working on the development of (multi-dimensional) separation strategies for the characterization of intact proteins.

Koen Sandra is Scientific Director at the Research Institute for Chromatography (RIC), which provides world-class chromatographic and mass spectrometric support to the chemical, life sciences and (bio)pharmaceutical industries. As a non-academic scientist, Sandra is author of over 40 highly-cited scientific papers and holder of several patents related to analytical developments in the life sciences area.

Shabaz Mohammed, after finishing his PhD in mass spectrometry, moved to Denmark to work with Ole Jensen in the field of proteomics and, in particular, the development of techniques for improving protein information. In 2008, Mohammed became group leader and Assistant Professor in Utrecht and worked with Albert Heck at the Netherlands Proteomics Centre. In 2013, he moved to the University of Oxford where is now Associate Professor.

John Yates III is Ernest W. Hahn Professor of Chemical Physiology and Molecular and Cellular Neurobiology at The Scripps Research Institute, LaJolla, California, USA. Yates was recently named Editor of the Journal of Proteome Research.

Where has 2D-LC had the largest impact?
SM: There are a significant number of examples demonstrating the power of multidimensional chromatography and it’s quite difficult to highlight a particular example. There are entire fields that depend upon it. Characterizing how signals are transported through a cell often requires an understanding of the behavior of proteins being phosphorylated. The presence and absence of this small molecule on proteins can determine if a protein is active, where it is in a cell, and with what it interacts. Phosphorylated proteins are low in abundance (occupying the lower levels of protein dynamic range) and such events are thought to number in the hundreds of thousands if not millions in a cell at any time. Identifying these events and their meaning often leads to multiple rounds of chromatography for enrichment and complexity reduction. AG: Offline 2D-LC has been widely used as a pre-fractionation strategy (prior or after protein digestion), to reduce the complexity of single shotgun RPLC-MS analysis. However, the long times required and big advancements in UHPLC-HRMS limited the spread of such workflows. An area where 2D-LC will continue to expand its impact is the selective analysis of part of the proteome, enriching certain protein or peptide species using affinity tags and/or special sorbents. KS: I have worked at a molecular diagnostics company for several years, where we were applying proteomics to discover and verify disease biomarkers. Using 2D-LC, we could mine the otherwise hidden proteome (hidden biomarker candidates), which came in particularly handy in the discovery programs. 2D-LC, yet in another format (targeted), was also successfully implemented in our biomarker verification workflows. Now that my focus has in recent years shifted more to biopharmaceutical analysis, 2D-LC based proteomics technologies again come in very handy to identify and quantify host cell proteins (HCPs). While in a 1D chromatographic set-up, the separation space is dominated by peptides derived from the therapeutic protein, the increased peak capacity governed by 2D-LC allows one to look substantially beyond the therapeutic peptides and detect HCPs at low levels (sub ppm relative to the therapeutic). We have also used 2D-LC in the quantification of therapeutic proteins in blood plasma to support (pre-)clinical development (pharmacokinetic studies), which is technically identical to biomarker verification. We have even validated these methods according to EMA guidelines. In these projects, the first dimension is used to reduce the matrix complexity prior to second dimension LC-MS analysis using multiple reaction monitoring. Because of matrix effects associated with 1D-LC-MS, one only obtains sensitivities in the high ng/mL range. Incorporating that extra dimension allows one to reach the low and even sub ng/mL levels in blood plasma/serum.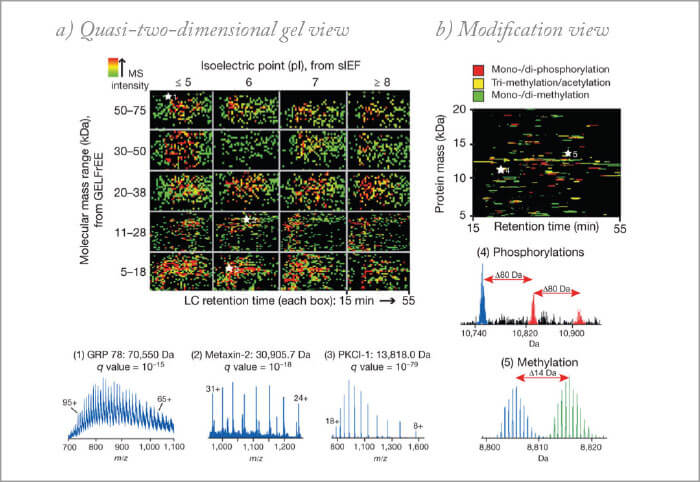
Are there any shortcomings to the technique?
SM: Coupling multiple rounds of chromatography is still not a trivial task. Multidimensional chromatography is still, mostly, used by committed analytical chemists. Sensitivity is of paramount importance in proteomic experiments. HPLC columns often lead to unacceptable losses unless attention is paid to how the sample is brought to each chromatographic system and how it is treated after fractionation. Miniaturization plays a huge role in proteomics and it is often not straightforward to reduce dimensions and flow rates while increasing column pressures. A significant amount of know-how about the various flavors of chromatography and the underlying science is required to pick and build multidimensional systems for certain types of proteomic experiments. That said, for unmodified ‘regular’ proteins, a consensus is being reached and so certain two-dimensional configurations have now hit mainstream science. AG: The major drawbacks of comprehensive 2D-LC (LC×LC) are: long analysis time (typically several hours), increased dilution (thus reduced sensitivity) in comparison with 1D-LC, and the complexity of method optimization. In analytical-scale separations, the introduction of systems with reduced dispersion volumes and UHPLC technology has drastically reduced the analysis time of LC×LC, enabling second dimension separations of about 20 seconds for a full gradient elution run. This is not yet the case for nano 2D-LC setups that are used for sample-limited applications, such as proteomic research. Here, due to dead and dwell volumes, the speed of the 2D cycle is typically limited to longer cycles (more than 10 min). In the coming years, further advancement in the miniaturization of LC apparatus (e.g. chip-based chromatography) will enable faster 2D separation cycles and thus facilitate the development of faster LCxLC applications. Several groups are working on the reduction of dilution in LC×LC and promising results are coming from the use of trap cartridges to collect fractions from the first dimension (what we call “active modulation”) and inject small volumes in narrow second dimension columns. Method optimization remains a challenge. However, software solutions to reduce the time and effort required to optimize two-dimensional methods will help analytical scientists in this task. KS: In addition to the technical challenges already mentioned, I believe one of the shortcomings is related to nomenclature. All kinds of different terms are being used to describe the way 2D-LC separations are performed (comprehensive, heart-cutting, LCxLC, LC-LC, off-line, on-line, automated off-line, and so on). In some ways, this is not surprising given that the technology has been developed from two different angles (proteomics and chromatography, respectively). In multidimensional GC, there are no ambiguities around nomenclature/terminology. People often contact us to ask which kind of 2D-LC they are actually performing. Importantly, depending on how 2D-LC is performed, one is confronted with flow and mobile phase incompatibilities, reproducibility issues, immature data analysis software, and so on. In the early days of using 2D-LC in proteomics, repeatability and reproducibility were not considered to be important issues. Now that the technology is really being applied to solve problems, old figures of merit have become primordial. JY: The biggest shortcoming is the time required to perform the analysis, which limits the number of experiments that can be performed. Let’s hope higher throughput methods for 2D-LC can be developed.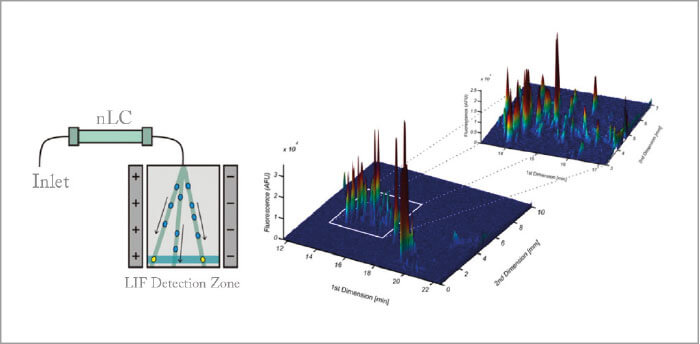
What is the current– and the potential – use of 2D-LC in proteomics?
AG: From what I have experienced, 2D-LC is used in specific studies where it is necessary to reduce the complexity and/or select part of the sample components of complex protein digests (bottom-up proteomics). Two-dimensional separation is also common in the analysis of intact proteins from cell lysates (top-down proteomics, see Figure 1). However, the longer analysis time and more sample handling steps of 2D-LC restrict its use in routine analysis of large numbers of samples. Some laboratories apply the Multidimensional Protein Identification Technology (MudPIT), introduced by John Yates’ lab at the beginning of this century. This separation method is the only online 2D-LC setup commonly adopted in proteomics research and many publications have demonstrated the advantages of its high separation power. Yet the longer analysis times and the fact that this setup is exclusively limited to ion-exchange and reversed-phase combinations have limited the spread of this technique. An interesting advancement of 2D-LC in proteomics may come from the development of online comprehensive two-dimensional chromatography (LC×LC), where independent chromatographic selectivities can be combined online, enabling more detailed analysis within times comparable to 1D-LC. Successful development in this area will greatly benefit research in top-down proteomics. KS: As Andrea suggests, MudPIT made the large scale analysis of proteomes possible. Personally, I find it remarkable that the 2D-LC technology was independently developed by two research communities, which can clearly be seen in the different ways 2D-LC is performed nowadays. On the one hand you have the proteomics people who are historically more MS driven (Yates) and on the other hand the chromatographers (Jorgenson). Chromatographers want to maximize resolution while proteomics scientists want to maximize the number of proteins (and eventually PTMs) that can be identified and quantified. The chromatographers, who typically did not develop the set-ups from a proteomics perspective, want to maximize resolution, which means that they have to maintain the first dimension separation upon transfer to the second dimension. To achieve that, peaks are sampled several times and stored in loops installed in between the first and second dimension column. Small internal diameter columns (1 mm or higher) are used in the first dimension and columns with wider internal diameter operated at high flow rates (mL/min) and high speed (< 1 min) in the second dimension. Indeed, from a theoretical perspective, this approach maximizes peak capacity (since first dimension separation is maintained upon transfer to the second dimension) but it is not at all compatible with mass spectrometry and proteomics. Indeed, flow rates in the mL/min range and second dimension run times below 1 min do not allow the detection of a substantial number of peptides. Multidimensional set-ups in proteomics typically use columns with small internal diameters in the second dimension (75 µm) that are operated in the nL/min flow regime. Second dimension gradients are also allowed to develop slowly (no real time constraints). As such, run times and flow rates are fully compatible with MS. The first dimension typically uses wider internal diameter so as to allow a high column load. Proteomics scientists do not worry about under-sampling the first dimension peaks. The number of samplings from the first dimension is typically low and the separated compounds are reunited, typically on an enrichment column. The two disciplines are evolving towards one another but, unfortunately, one now sees studies/papers appearing from chromatographers that were, in fact, already described 10 years ago in specific journals by proteomics scientists. SM: The idea of 2D-LC as the best means to obtain a comprehensive proteome is pretty much accepted by the entire field. The exciting thing is that all the know-how built up over the last few decades now means that there is a much better awareness in the field on how to perform a 2D-LC experiment. Manufacturers are now providing far more appropriate and, more importantly, robust components to build 2D-LC systems. I think separation power will continue to improve but my expectation is that 2DLC will become the main approach ahead of one dimensional LC-MS for obtaining a comprehensive proteome analysis. JY: The method is pretty well established as an off-line technology. You can also view methods such as IMAC in combination with 1D-LC as a 2D-LC method. I would say that most of the uses of 2D-LC are off-line. These methods are driving a lot of discovery in academic laboratories as the capability for more comprehensive discovery grows.Who has played large roles in 2D-LC development, technically as well as theoretically?
AG: I have only been involved in 2D-LC research over the last three years. In this time my work has greatly benefitted from theoretical studies and technological developments resulting from the vison and efforts of many researchers. Certainly major developments have been driven by analytical “shrines” such as Jorgenson, Carr, and Yates’ labs that practically realized online 2D-LC and participated in the development of the instruments that are now on the market. KS: I agree with Andrea that there are certainly notable pioneers, such as Jorgenson (chromatography) and Yates (proteomics), but many others have substantially contributed as well – as with all questions of this type, it is too risky to forget important contributors with a definitive list. I would say many of the major vendors, including Dionex (now Thermo Fisher Scientific, but early developments took place at LC Packings/Dionex), Waters and Agilent are also at the forefront. SM: As Koen notes, the number of people involved in 2D-LC development is far too long to list, although one of my fellow interviewees played a major role. John Yates’ work on 2D-LC combined with his work on ‘shotgun’ proteomics helped lead to a sea change in how the field went about protein characterization. The importance of 2D-LC is such that all major LC and MS manufacturers are invested in active research and development in chromatography. One of the first dedicated solutions for nanoflow LC (a staple of proteomics) was developed by a little company called LC Packings/Dionex that, after a number of takeovers, became part of Thermo Fisher Scientific, as Koen says. JY: Long before we tried 2D-LC for proteomics, I followed mostly the work of Jorgenson in this area. I was always impressed with the elegance and potential of the method. I know many others contributed to this area, but I was following the mass spectrometry and protein sequencing areas so my bandwidth was limited. Jorgenson was always pushing the outer limits of separations like CE at million volts, LC at 100,000 psi so his research was always “Gee Whiz” kind of stuff. It was fun to see what he would think of next!What’s the most important 2D-LC development of the last ten years?
SM: Tough question. It’s been a very smooth evolution rather than a revolution. But the moment that sticks in my mind was when there was a collective realization that 2D-LC was the best way to obtain deep proteome characterization, which then led to many different chromatographic combinations. The need to find optimal complementarity led to new flavors of chromatography as well. KS: For me, it’s the move away from specialized labs using home-built systems to use by a broader audience, which has been facilitated by the introduction of commercial instrumentation. AG: The use of UHPLC technology in two-dimensional separation systems following the research lead by Stoll, Carr and colleagues that resulted in the launch of robust systems capable of second dimension separation performed in cycles of 20 seconds. JY: For me, the most important development has been combining 2D-LC with ultra-high pressure LC. The commercial availability of high pressure LC pumps opened up the use of this technique to a broader array of people and that helps to drive further innovation.Where does multidimensional separation go from here?
AG: I’d like to think that in the future multidimensional systems won’t exclusively be coupled in the time domain (where sample fractions are collected from a first separation and each of them is consecutively analyzed by a second separation) but will also use spatial dimensions. The possibility and power of this approach was shown recently by Bowser et al, coupling RPLC (time) with free flow electrophoresis (space) achieving an impressive peak production rate of 105 peaks/min (see Figure 2). More approaches to realize spatial × time based chromatography may arise from efficient devices for planar chromatography. Whatever form multidimensional separation takes, I look forward to highly sophisticated technologies that are fast and easy to use! KS: Again speaking from a proteomics perspective (if you asked me the same question from a non-proteomics perspective, I would give a completely different answer!) I believe that both (or more) dimensions will be integrated in chip-like devices that act as sample introduction systems for MS. Once more, what is required in proteomics cannot be generalized, which is to say, low volume in the second dimension. SM: Our understanding of manufacturing materials and refining selectivity is improving at a rapid rate. Our ability to operate at ever-higher pressures with columns packed with increasingly small particles is reaching an astounding pace. The performance of LC must go hand in hand with improvements in MS speed for the field of proteomics, but I am not concerned about that as an issue. I agree with Koen that there is a strong possibility that 2D-LC can be made modular and perhaps in chip form – at that point, the market will massively increase. Miniaturization and robustness is certainly the future. JY: Put simply, it must be faster with greater peak capacity.Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.




