Just as proteins are the third component in the flow of genetic information after DNA and RNA, so proteomics represents the third challenge temporally in the comprehensive analysis of living systems, after genomics and transcriptomics. It is also the most complex of these challenges. Proteins are much more diverse and difficult to quantitate than nucleic acids, and unlike DNA, their expression varies in both time and space.
Knowledge of protein sequence and abundance has many important roles in biotechnology and medicine. One major application is biomarker discovery, for which the availability of patient samples is an essential resource. The hypothesis behind biomarker studies is that a disease, such as cancer, leaves tell-tale markers in serum that can be used for diagnosis and prognosis. Perhaps the best-known marker, prostate specific antigen (PSA), illustrates the complexity of the field. PSA was developed as a prognostic indicator for patients after treatment for prostate cancer. Its subsequent widespread use as a screen for the general population was not adequately validated and is now widely discouraged. Proteomics is also used to identify cellular networks that are deregulated in disease. Such studies compare primary tissue from diseased and healthy individuals, and the goal is to identify potential therapeutic targets. Another application is the quality control of recombinant therapeutics. These protein-based drugs are produced in cell culture. The host cell, which is often the Chinese hamster ovary (CHO) cell line, is engineered to produce the desired therapeutic. The cells or cell culture is then harvested, lysed, and taken through a set of affinity purification steps. However, residual proteins from the host cell line remain, albeit at trace levels. The host cell proteins represent potential allergens, and it is vital to both identify the contaminants and determine their abundance. One class of recombinant therapeutics is antibodies, which are generated from polyclonal antibodies against the target molecule. Antibodies represent the exception to the rule that genomic sequence informs protein sequence within an organism. The complex immunological system shuffles the sequence for antibodies, so that each immunological cell contains a unique genetic sequence for the antibodies that it produces. As a result, it is necessary to perform de novo protein sequence determination for target antibodies. Finally, post-translational modifications decorate proteins with a wide range of functional groups. These functional groups play a diverse role in modulating a protein’s function. Phosphorylation is the most commonly studied post-translational modification because of the availability of robust tools, the well-understood role that phosphorylation plays in modulating protein function, and the simple nature of the modification. It is not uncommon to see studies where the phosphorylation status of a large fraction of the proteome is studied. In contrast, another major post-translational modification, namely glycosylation, has received much less attention. This lack of attention does not reflect a lack of importance, but rather the complexity of the modification and the primitive status of analytical tools.
Although it is considered to be the younger sibling of genomics, protein sequence analysis is in fact older than DNA sequencing. It originates in the late 1940s/early 1950s with Sanger’s sequence determination of insulin and Edman’s development of the isothiocyanate degradation reaction. Those technologies are labor intensive, slow, and require large amounts of starting material. It was quite rare to determine the entire sequence of a protein by use of Edman’s chemistry. Instead, that primitive technology was used to generate the sequence of a small peptide created from the protein, perhaps consisting of a dozen or so amino acids. The genetic code was then used to create probes for the corresponding gene. Libraries created from fragments of the genome would be screened for the target gene, which would then be sequenced. As we described in the inaugural issue of The Analytical Scientist, genomics underwent explosive growth and maturation over the past twenty years. One legacy of this is the complete genome sequence of a very large number of organisms. That genetic information, in turn, has been used to populate databases with expected protein sequences for those organisms. It is no longer necessary to synthesize oligonucleotide probes corresponding to the sequence of a peptide and then screen a library of genetic fragments to identify the gene of interest. Instead, the peptide sequence is screened in silico against the genetic sequence. The availability of complete genomes has been an extraordinarily valuable resource in proteomic studies.
To maximize the potential of connecting genetic and protein sequences, efficient methods for generating sequences from short peptides was needed and in the 1970s mass spectrometry was identified as the ideal tool. The main challenge was to get large, rather non-volatile proteins or peptides into the gas phase for ionization and analysis. A number of approaches were investigated, including ultrasonic nebulization and secondary ionization mass spectrometry. These were superseded by the development of electrospray ionization by Fenn and MALDI (Matrix-assisted laser desorption/ionization) by Tanaka and Hillenkamp in 1988. Together, these approaches opened up the use of mass spectrometry for high-throughput proteomic studies. Three additional advances were required for widespread proteomic studies. First, as we noted in our article in the January issue of The Analytical Scientist, genomic sequence databases approached reasonable size in the 1990s. Since then, whole-genome sequencing studies for many organisms have populated databases with the entire cast of protein sequences. Second, beginning with the pioneering effort of Yates and colleagues around the year 2000, the development of efficient database search engines automated the identification of those protein sequences. At this point, we knew what we were looking for. And third, beginning with the iTRAQ (isobaric tags for relative and absolute quantitation) chemistry of Aebersold and colleagues in 2000, a suite of reagents and tools was developed to undertake quantitative proteomics, enabling us to understand changes in protein abundance that accompany disease and development. Proteomic studies are commonly divided into top-down and bottom-up (see Figure 1). Top-down proteomics ionizes and introduces intact proteins into a high-resolution mass spectrometer. This approach has the advantage of capturing all posttranslational modifications within the protein but suffers from the disadvantage of needing to ionize, fragment, and interpret the resulting data for a very large molecule.
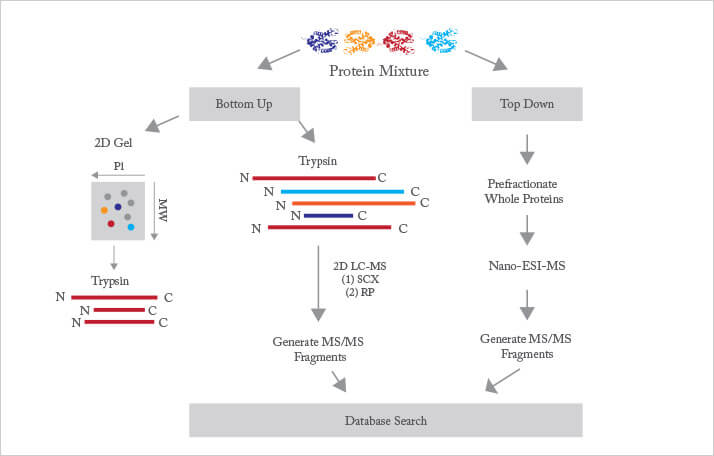
The vast majority of proteomics is based on the bottom-up protocol, where the complex mixture of proteins from a cell, tissue, or organism is enzymatically digested into a very large mixture of peptides. Trypsin is the proteolytic enzyme of choice. This enzyme is available at high purity and is relatively inexpensive. It cuts proteins at lysine and arginine residues. Since these amino acids are relatively common, each protein is cut into many peptides, which are typically five to 30 residues in length. While this mass range suffers from significant interference from the matrix used in MALDI, it is very well suited to analysis by electrospray ionization, which is most commonly used. Determination of most of the peptide sequence, along with knowledge of the parent ion’s mass, narrows identification to a small number of possibilities in database searches. Early proteomic studies were focused on identifying proteins within a given sample, essentially providing a parts-list of the proteins present. A modicum of quantitative information can be gleaned from such a parts list, most commonly by simply counting the number of occurrences of peptides from a target protein. More recently, differential isotopic labelling has been used to determine changes in protein abundance between two or more samples. For example, protein abundances from normal and diseased tissues can be compared to identify enzymatic and signalling pathways associated with the disease. Modern bottom-up proteomic studies consist of four stages. First, the protein samples are digested and isotopically labelled. Second, the resulting complex peptide mixture is subjected to one or more rounds of separation. Third, the sample is ionized and analysed by the mass spectrometer. Fourth, the resulting data is analysed and interpreted. Below, we consider recent advances in each of these four areas.
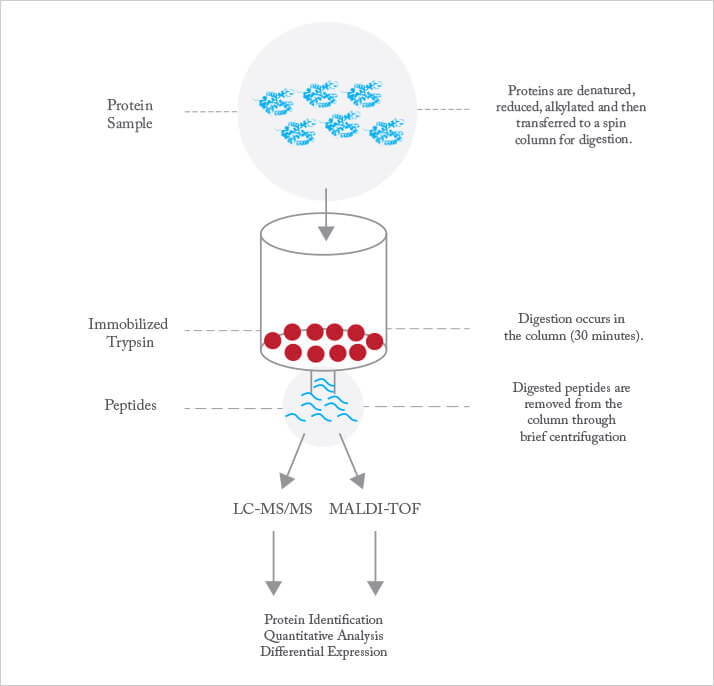
Trypsin digestion is performed for anything between 30 minutes and 24 hours. It’s a trade-off: digestion periods at the shorter end reduce the analysis time and minimize auto-digestion, but at the expense of incomplete digestion, which results in missed cleavages, while longer digestion time leads to more complete digestion but at the expense of more auto-digestion and longer overall analysis time. Trypsin may be immobilized on a solid support, which dramatically increases the concentration of enzyme used in the reaction while minimizing auto-digestion of the enzyme (see Figure 2). Immobilized trypsin provides roughly an order of magnitude improvement in sample throughput, which is of value in, for example, control of recombinant therapeutic protein production. Speeding digestion is particularly important in the bio-pharmaceutical industry, where rapid proteomic analysis is vital for quality control of therapeutic proteins produced in cell cultures. The goal is to provide information rapidly enough for it to be used for process control.
The mixture of peptides present in the tryptic digest of a complex proteome is far too complex for direct analysis by mass spectrometry. Instead, the mixture must be separated to simplify the sample sprayed into the mass spectrometer. In the overwhelming majority of cases, this separation is achieved by gradient-elution reversed-phase liquid chromatography. This separation mode employs solvent systems that are compatible with electrospray ionization, and that provide outstanding separation resolution. The resolution of chromatographic separations improves dramatically as the size of the stationary phase particles decreases. However, the pressure required for the separation also increases as particle size decreases. The development of technology for routine ultrahigh pressure liquid chromatography has resulted in remarkable improvements in the separation of proteolytic peptides for mass spectrometric analysis.
Virtually all automated proteomic analyses employ liquid chromatography to separate peptides before analysis. However, there are hints that capillary electrophoresis may prove of value in proteomic analysis. We have developed a novel electrospray interface, which uses extensive sample pre-fractionation in combination with rapid capillary electrophoretic separations; combined with high-speed and high-resolution mass spectrometers, this provides an intriguing alternative. In the largest published comparison of capillary electrophoresis with UPLC separation, the two techniques achieved an essentially identical number of protein identifications in an identical analysis time. In this analysis, which was the study of a bacterial secretome, the two techniques had roughly fifty percent overlap in the proteins that they identified, suggesting that they probe complementary portions of the proteome. As an aside, this study also demonstrates that no single technique is capable of resolving all components within a complex proteome, and that many situations will benefit from combination of two or more techniques.
The development of high-speed and high-resolution mass analyzers has revolutionized proteomic research. One class of instruments, the Orbitrap, is quite striking in its performance (Figure 3). These instruments routinely achieve 100,000 mass resolution (at m/z = 400), produce tandem spectra at 10 Hz, and achieve low attomole detection limits for parent ions.
This area of research is very competitive, and new generations of mass spectrometer appear every three to five years. We will undoubtedly see continued advances in mass analysis for many decades.
Quantitative proteomics
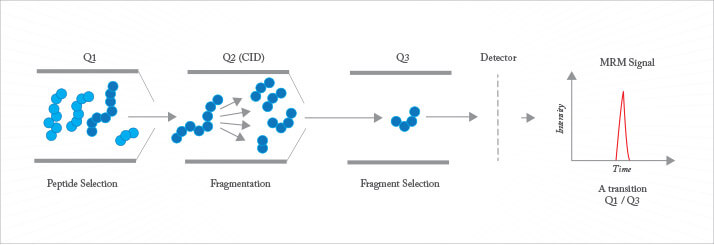
Early proteomic studies delved deep into the proteome, with the goal of detecting a large fraction of the proteome predicted from the translation of open reading frames in the genome. However, such parts lists are of limited value, since they do not directly provide information on the change in abundance of proteins between samples, such as those from healthy and diseased tissue. A number of strategies have been developed for quantitative proteomics, with many more under development. Most methods employ isotopic labelling of primary amines found on lysine residues or the N-terminus of peptides, or sulphurs on reduced cysteine residues, using specific reagents. Two or more samples are treated with different isotopic labels, the samples are then pooled and analyzed using conventional chromatographic and mass spectrometric techniques. Software is used to identify the isotopic signature from the mass spectra. Reagents can be as simple as formaldehyde, which introduces an isotopic signature, to the iTRAQ chemistry that employs reagents that reveal the isotopic signature only after fragmentation during tandem mass spectrometry. Alternatively, label-free methods can be employed to quantify protein abundance. These include spectral counting, where the number of peptides identified for a protein provides a surprisingly simple estimate for rough quantitation. Much higher precision is provided by multiple reaction monitoring (MRM), also known as selected reaction monitoring (SRM) (see Figure 4). This approach employs a triple quadrupole or a Q-trap instrument. The device is programmed to isolate a specific parent with the first quadrupole, to fragment that ion in a low-pressure cell, and then monitor the intensity of a specific fragment ion. MRM/SRM provides up to four orders of magnitude linear dynamic range and sub-attomole detection limits. It is only useful when the target peptide has been identified and when that information is available for construction of a calibration curve.
As mentioned previously, most proteins have functional groups added after translation. These modifications play a vital role in determining the protein’s function, cellular location, and lifespan. Three hundred different types of post-translational modification have been described, of which only a few have been studied in detail at the proteome level. Phosphorylation analysis has received the most attention, in part because of the importance of phosphorylation in protein function but also because of the relative simplicity of the analytical challenge. Most phosphoproteomic analyses employ affinity reagents, such as metal ions held on a stationary phase (the technique is known as ion mediated affinity chromatography, or IMAC), to selectively enrich phosphorylated peptides from tryptic digests. The captured peptides are eluted and analyzed by liquid chromatography coupled with tandem mass spectrometry. Analysis of the fragmentation spectrum is used for high-throughput identification of phosphorylation sites. Despite the many advances made in phosphorylation analysis, challenges remain. Highly acidic peptides also are captured by IMAC columns and, perhaps more importantly, phosphate groups can cleave or migrate during fragmentation: both of these confound the analysis. Current techniques excel at identifying sites of phosphorylation but are less useful in ascertaining the extent of phosphorylation at a particular site. The main issue is that the phosphorylated and unphosphorylated versions of a peptide vary in their charge, and hence in their ionization efficiency. Calibration of the ions’ responses is a challenge for high-throughput studies.
Glycosylation, an equally important post-translational modification, is much more challenging for current analytical tools. While glycosylation modifications can be quite simple, for example, consisting of a single sialic acid, glycans are produced in a bewildering variety of structures. Their analysis usually proceeds by enzymatic cleavage of the glycan from the peptide, followed by mass spectrometric analysis. This has two limitations: first, the relationship between the glycosylation site and the glycan’s structure is lost; and second, mass spectrometry has challenges addressing isomeric structures.
Proteomics is at a much earlier stage of development, and represents a much greater analytical challenge, than genomics. We estimate that proteomics will require two to three decades of development before quantitative analysis of whole proteomes, including post-translational modifications, becomes routine. While it is now relatively straightforward to delve deeply into a proteome, doing so for quantitative analysis remains time consuming and expensive. The identification of post-translational modifications, and determination of the relative abundance of those modifications within a single sample, present complex challenges. One area of promise is in situ proteomics. It is now possible to visualize the microscopic distribution of metabolites and small peptides using MALDI imaging (see Figure 5).
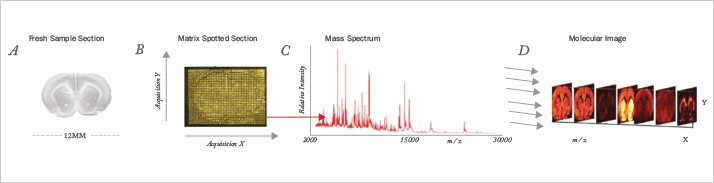
However, performing proteomic analysis with high spatial fidelity across a tissue is a formidable task. Among the issues are the uneven deposition of MALDI matrix on the tissue and the use of a relatively large laser beam that averages the signal across several cells. Other challenges include the identification of detected ions and the modest sensitivity that the approach has. Other factors that dog proteomics are superficially trivial, yet remain unsolved. For example, there are huge banks of archived human tissues available across a range of diseases but most of these samples are intractable for the current generation of analytical tools. Samples are generally fixed in formalin or frozen. Formalin cross-links primary amines, and these cross-links are difficult to reverse without sample loss and damage. And many of the frozen tissue banks use OCT (optimal cutting temperature) media. This contains large amounts of polyethylene glycol, which generates a complex mass spectral background signal that interferes with the analysis of tryptic digests. Top-down proteomics – the analysis of intact proteins – simplifies a number of steps in protein analysis. However, it requires very high-resolution instruments and large-abundance protein samples. Advances in instrumentation, sample separation, and data analysis are required to make top-down proteomics a routine tool. As we noted at the beginning of this manuscript, proteomics plays an important role in several areas of pharmaceutical research. First, differences in protein expression between healthy and diseased tissues provide targets for drug development. Second, the preparation and quality control of recombinant therapeutics requires proteomic techniques with a wide dynamic range to identify impurities in the presence of high abundance therapeutics. Finally, the characterization of protein abundance variance during development and disease progression will provide a deep understanding of basic biology in health and disease.
Newsletters
Receive the latest analytical science news, personalities, education, and career development – weekly to your inbox.